Understanding Solvent Effects in Catalysis

Van 't Hoff Institute for Molecular Sciences and Amsterdam Center for Multiscale Modeling, University of Amsterdam, Amsterdam, The Netherlands
e-mail: e.j.meijer uva.nl (e[dot]j[dot]meijer[at]uva[dot]nl)
uva.nl (e[dot]j[dot]meijer[at]uva[dot]nl)
Rational design of novel efficient catalysts requires an understanding and quantitative picture of the mechanism, energetics and kinetics of the catalytic cycle. In our approach1,2, we address this by using density functional based molecular dynamics (DFT-MD) methods, taking the role of temperature and solvent explicitly into account.
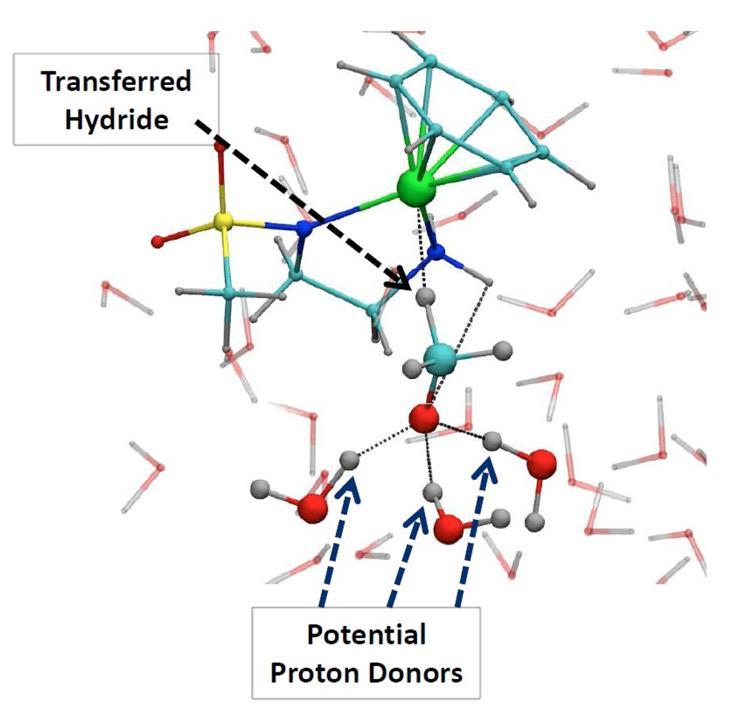
We demonstrate how, for important types of metal-based molecular catalyst, this approach can provide novel fundamental insight. In particular we show how protic solvent molecules can play an active role by participating via hydrogen bonding and mediating proton-transfer processes. This yields a picture of the free-energy profile, reaction mechanism and kinetics that can be fundamentally different from that predicted by static gas-phase calculations without explicit solvent.
References
[1] J.-W. Handgraaf and E.J. Meijer, J. Am. Chem. Soc. 129, 3099 (2007); A. Pavlova and E.J. Meijer, ACS Catal. 6, 5350 (2016).
[2] V. Sinha, N. Govindarajan, B. de Bruin, E.J. Meijer, ACS Catal. 8 6908 (2018); N. Govindarajan, H. Beks, E.J. Meijer, ACS Catal. 10, 14775 (2020).