1. Introduction
Catalysis is generally associated with underpinning approximately 30% of gross domestic product in European economies.1 Catalysis is involved at some point in the processing of over 80% of all manufactured products.1 Zeolites compose the most important group of heterogenous industrial catalysts. The need of (i) the transformation of the chemical production to a sustainable one and (ii) the decarbonization and transfer to new substrates requires the development of a new generation of heterogeneous catalysts, especially zeolite–based ones. Zeolites are very widely applied due to their (i) enormous tunability allowing a high activity and selectivity, (ii) mechanical and chemical stability, and (iii) excellent transport properties and accessibility of reaction centers. The design of advanced catalytic systems exhibiting high activity and selectivity and meeting requirements of industrial applications represents a complex process that cannot be performed without the detailed knowledge of the properties of the catalysts.
Zeolites are crystalline microporous aluminosilicates [Sin-mAlmO2n]m- made of corner sharing TO4 tetrahedra (T = Si and Al-; Si in SiO4 and Al- in AlO4- are isoelectronic). There are two types of zeolite atoms – framework ones and extra–framework species. The former form the framework of a zeolite while the latter which are positively charged coordinate to framework O atoms of AlO4- tetrahedra to compensate the negative charge introduced by framework Al- atoms. A typical feature of many silicon–rich zeolites is a high number of crystallographically distinguishable T (i.e., tetrahedral) sites. Since the protons, cations, and metal–oxo species (i.e., positively charged extra–framework species) bind to O atoms of AlO4- tetrahedra, the crystallographic position of aluminum in the zeolite framework governs the location of the active sites, which in turn affects the catalytic activity and selectivity.2
The organization3 of Al atoms in the framework of Si–rich zeolite catalysts is a key property.3-6 The Al organization3 includes (i) the Al siting (i.e., which different crystallographically distinguishable framework T sites are occupied by various types of Al atoms),7 (ii) the Al distribution (the distribution of framework aluminum atoms among various types of Al atoms),3 and (iii) the location of framework Al atoms of interest in the channel system of the zeolite.8 The positively charged active species balance the negative charge of AlO4- tetrahedra, and therefore, the organization of Al atoms in the zeolite framework controls the formation and properties of active sites in the zeolite.4-5, 8-11 The Al siting determines the position of the active sites in the zeolite framework while the Al distribution controls the concentration and stability of mono and divalent cations and metal–oxo species.3-5, 12-21 In addition, for monovalent cationic species including protons, the Al distribution also controls the distance between the active sites and thus a possibility of their cooperation.22-23 Obtaining insights into the Al organization is of crucial importance for the development of new better catalysts as catalytic studies showed that zeolites of the same chemical composition but different Al organization could possessed different catalytic properties.4-6, 8, 24-25 Thus, the potential of a zeolite for individual catalytic reactions cannot be evaluated without the knowledge of the Al organization in the framework.
Diffraction techniques cannot distinguish between Al and Si atoms in the zeolite framework, and therefore, do not allow direct identification of the Al siting in zeolites.2, 26-27 The Al siting of Si–rich zeolites with several crystallographically distinguishable T sites had not been known before 2007. We developed the new bare framework model2, 26-30 which includes neither water molecules nor explicitly counter cations and used it in our DFT calculations in tandem with 27Al (3Q) and 29Si MAS NMR spectroscopy and determined for the first time (i) the partial Al siting in a set of ZSM–5 zeolites2, 26, 29 and (ii) the full Al siting in a set of ferrierite.27 The interpretation of the 27Al (3Q) NMR spectra would not have been possible without our DFT calculations. Our developed methodology to determine the Al siting was subsequently used by others.31-32
In addition, we showed for the first time that monovalent cations (e.g., Li+ and Na+) in dehydrated zeolite frameworks could be used as probes of the Al siting.17, 21 However, even in this case diffraction methods could not be employed to study Li+ and Na+ centers in Si–rich zeolites because of a large number of possible cationic sites and a low or no occupancy by the cation of these sites due to a low content of the cation in the zeolite.7, 17, 21 Therefore, we developed a new methodology using high–resolution multinuclear 7Li and 23Na solid–state NMR spectroscopy coupled with DFT calculations and showed that this methodology represents a powerful tool to identify the corresponding siting.17, 21
Li+ cations (monitored by 7Li MAS NMR spectroscopy coupled with DFT computations) employed as probes of the Al siting can serve as a complementary method to 27Al (3Q) MAS NMR spectroscopy. Li+ cations are very likely the most useful probes among monovalent cations because they are small and strongly coordinate to one or two framework oxygen atoms of AlO4-.17 Na+ cations monitored by 23Na (3Q) MAS NMR in tandem with DFT calculations can determine the ring forming the Na+ site but not which T site is occupied by Al in that ring.21
Besides Brønsted acid SiOHAl groups formed by protons compensating tetrahedral AlO4-, also electron–pair acceptor Al Lewis sites are often present in zeolite catalysts.33-35 The Al Lewis sites were suggested to correspond to Al centers tricoordinated to the zeolite framework.34, 36-37 However, this type of Al has resisted detection by 27Al MAS NMR till our investigation.38 Our subsequent solid–state NMR and DFT study39 for the first time (i) showed that the electron–pair acceptor of the Al framework (AlFR) Lewis sites corresponded to an AlTRI atom tricoordinated to the zeolite framework which adsorbed a water molecule and (ii) proposed a plausible mechanism of the formation of (AlFR) Lewis sites in the beta zeolite.
In contrast to the Al organization, the siting and location of bare divalent cations M(II) in the rings and the channel systems, respectively, were known9-11 for a number of Si–rich catalysts analyzed by experimental methods – UV–vis combined with FTIR spectroscopy of Co(II) cations. However, obtaining the local structure and stability of these sites would have required the knowledge of the Al siting and even with this it could have been elucidated only by DFT calculations. Therefore, we developed a new procedure to obtain the local structure and stability of cationic sites formed by bare divalent cations accommodated in 6–rings and 8–rings. We discovered that periodic DFT calculations including molecular dynamics simulations or other similar global optimization techniques must have been used.3, 15, 18, 20, 40-45 We showed that the accommodation of bare divalent cations in rings forming cationic sites could have led to significant rearrangements of the local structures of the zeolite framework, and therefore, the precise local structure of sites binding a divalent cation could not have been derived from results of X–ray crystallography and neutron diffraction crystallography experiments, but could have been inferred from theoretical calculations.3, 15, 18, 20, 40-45 The calculated structure of the M(II) sites represents a starting point for the investigation of the performance of the cations in catalysis. Moreover, in zeolites with highly complex structures, the empiric interpretation of the spectroscopic experiments is not sufficient even for the analysis of the Me(II) siting in the ring and the channel.20 Our newly developed methodology was applied for the analysis of industrially important zeolite catalysts for the first time.3, 15, 18, 20, 40-45
Distant binuclear cationic sites were firstly identified using theoretical modeling in the context of the study of the N2O decomposition over the Fe(II) cation exchanged ferrierite, the beta zeolite, and ZSM–5 (i.e., Fe–ferrierite, Fe–beta, and Fe–ZSM–5, respectively).46 We devised that the presence of the active sites formed by the distant binuclear Fe(II) centers explained the exceptional activity of Fe–ferrierite in comparison with the Fe–beta and Fe–ZSM–5 catalysts.40 The first chemical step of the N2O decomposition is the formation of the α–oxygen species40-41 [i.e., (Fe(IV)═O)2+] which exhibits unique oxidation properties reflected in an outstanding activity in the oxidation of methane to methanol at room temperature.41, 47-51 Furthermore, we predicted for the first time employing periodic DFT calculations and subsequently confirmed experimentally that the ferrierite zeolite exchanged with other transition metal cations able of the M(II) to M(IV) redox cycle could be employed for the preparation of the α–oxygen species [i.e., (M(IV)═O)2+] using N2O.41
Moreover, we firstly predicted using the power of periodic DFT calculations that these distant binuclear cationic sites were able to split dioxygen to yield pairs of the distant α–oxygen species.43 Subsequently, experiments were performed at room temperature and the theoretical prediction of a cleavage of dioxygen to give a pair of the distant α–oxygen atoms was confirmed experimentally and thus splitting dioxygen was discovered.43 A pair of the formed distant α–oxygen species [i.e., (Fe(IV)═O)2+] features exclusive oxidation properties manifested in an exceptional activity in the oxidation of methane to methanol at room temperature.43 Theoretical modeling further clearly showed that this breakthrough52 in the activation of dioxygen was not limited exclusively to Fe(II) cations but the ability of dioxygen splitting represented a general property of the distant binuclear M(II) centers capable of the M(II) to M(IV) redox cycle with the Me...Me distance of ca 7.4 Å stabilized in M(II)–ferrierite.44 These findings were afterward verified experimentally.53 In addition, our computational study revealed that the distant binuclear Fe(II) sites with suitable parameters accommodated in various zeolites can split dioxygen to form a pair of the distant α–oxygen species as well.45 This outcome is most likely true also for other M(II) cations capable of the M(II) to M(IV) redox cycle. Therefore, the ability to cleave dioxygen represents a general property of the distant binuclear M(II) centers stabilized in aluminosilicate matrices, and thus suggesting the possibility of developing M–zeolite–based systems for the dioxygen activation for direct oxidations using various zeolite matrices.45 Afterward, our DFT study revealed that the proximity of the other (Fe(IV)═O)2+ site in the confined reaction space of the zeolite cavity could dramatically change the behavior of both the cooperating α–oxygen atoms and the reaction mechanism over (Fe(IV)═O)2+ sites of a pair of the distant α–oxygen atoms could differ from that over isolated (Fe(IV)═O)2+ sites.54
This Thesis describes the developments and results regarding the theoretical part of the determination of the Al organization including the key development of the bare framework model which allowed the simplification of the computational model to calculate reliable NMR parameters for the zeolites of interest. This permitted the achievement of the determination of (i) the partial siting of isolated single Al atoms in a set of ZSM–5 zeolites and (ii) the full siting of both isolated single Al atoms and Al–O–(Si–O)2–Al sequences in a set of ferrierite zeolites including the ferrierite sample used to prepare the distant binuclear cationic sites. The Thesis further reveals the local structure and dynamics of cationic sites for bare divalent cations in various zeolites. All the mentioned knowledge enabled our identification of the distant binuclear Fe(II) sites using theoretical modeling. These centers are responsible for the facile N2O decomposition in the Fe–ferrierite catalyst. Employing the power of periodic DFT calculations we predicted that these distant binuclear cationic sites could split dioxygen at room temperature to yield pairs of the distant α–oxygen species able to oxidize methane to methanol at room temperature. Based on this theoretical prediction, we discovered a cleavage of dioxygen over distant binuclear cationic sites employing Mössbauer and FTIR experiments and stoichiometric reaction tests. This achievement represents a breakthrough in oxidation catalysis.52 The obtained knowledge regarding the Al organization of the zeolites of interest and concerning the local structure of M(II) cationic sites was a necessary condition to discover splitting dioxygen on distant binuclear Fe(II) cationic sites, and subsequently, to prepare the α–oxygen atoms (using N2O or O2) on Fe(II) and M(II) cations other than Fe(II).
2. Zeolites5
Zeolites represent a wide and extremely important group of industrially used heterogeneous catalysts. They are used in a broad range of acid–catalyzed reactions for the transformation of hydrocarbons and their derivatives that are relevant for the petrochemical industry, as well as in the synthesis of fine chemicals.36, 55-56 Cationic zeolite forms are used as redox catalysts for NOx elimination from diesel exhausts and process gases and for N2O abatement.57-59 Moreover, zeolites in the protonic and cationic forms were recently reported to be promising catalysts in the utilization of biomass and renewables,60-62 in the conversion of methane into valuable products, and in the utilization of carbon dioxide.63-68
The enormously wide application of zeolites in catalysis results from a unique combination of the properties of zeolites. Their microporous crystalline aluminosilicate frameworks are composed of corner–sharing TO4 tetrahedra (T = Si, Al). Variability in the arrangements of the TO4 tetrahedra results in more than 200 known zeolite topologies with different microporous channel and cavity systems.7 Thus, zeolites assume the role of chemically, thermally, and mechanically stable matrices that are also rigid and well–defined at the atomic scale. Moreover, zeolites represent a highly variable microporous system with tunable properties. Framework Al/Si substitutions introduce a negative charge to the silicate framework. This negative charge has to be compensated by extra–framework cationic species: protons, metal, and metal–oxo cations. These exchangeable, positively charged extra–framework species can act as catalytic and sorption centers. The extremely wide range and also variable nature of extra–framework cationic species (i.e., Brønsted and Lewis acids, various redox and base centers)4, 69-70 acting as active sites in tandem with the tunable geometry and the architecture of the pore structure result in an enormously broad array of possible zeolite applications in catalytic processes. The structure of the pore system is responsible for the shape selectivity controlled by the transition states and the transport of the reactants and products through the pores. As mentioned above, the active sites balance the negative charge of the AlO4- species in the zeolite framework. Therefore, the organization of Al atoms in the zeolite framework determines the location, distances, properties, and nature of the active sites in the zeolite catalysts.
3. Cationic sites in pentasil zeolites27
Bare divalent metal cations can occupy in pentasil zeolites (e.g., mordenite, ferrierite, ZSM–5, and the beta zeolite) three types of cationic sites designated as α, β, and γ.9-11, 71 The presence of two Al atoms in the site is necessary for stabilization of bare divalent cation.12-13, 72 These three sites were determined using X–ray diffraction (XRD) for mordenite (sites E, A, and C according to the Mortier notation)73-74 and suggested employing UV–vis spectroscopy of bare Co(II) ions as a probe for ZSM–5,11 the beta zeolite,71 and ferrierite10 for which the sites were later confirmed by synchrotron powered XRD.75-77 Figure 1 shows the structure of the α and β sites while the γ site is not depicted since the concentration of framework Al atoms forming the γ site in ferrierites is very low.10
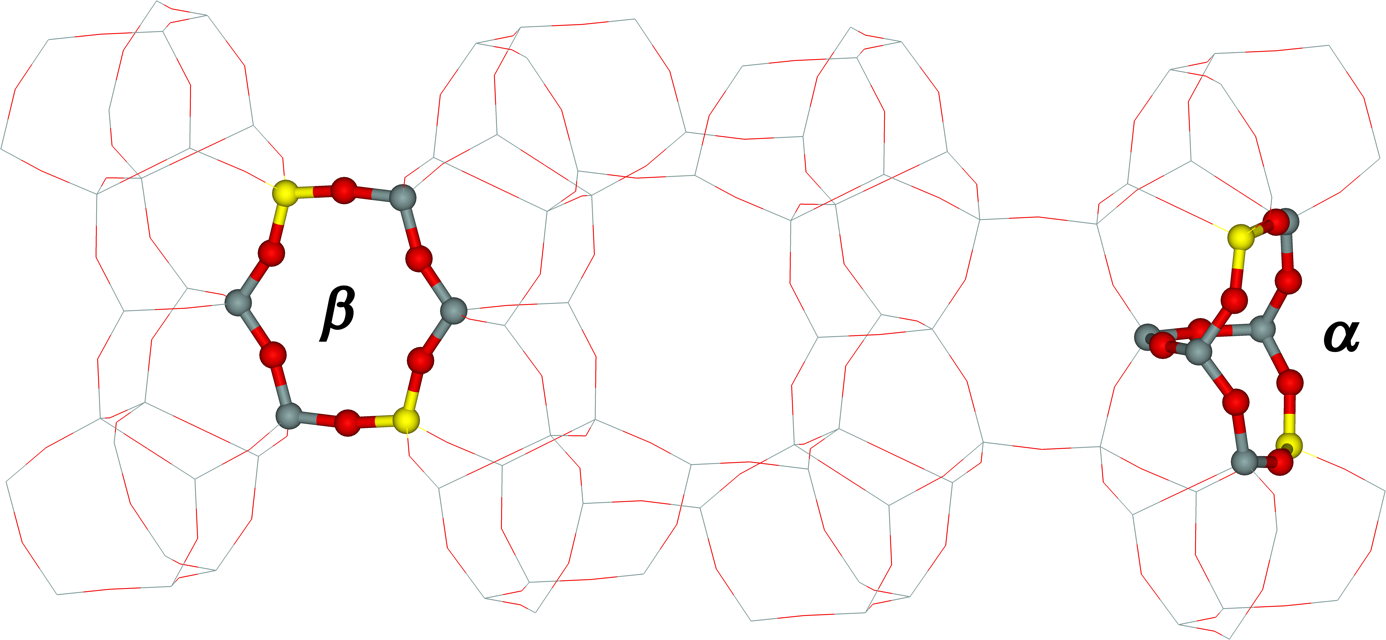
Figure 1. Structures of the β (left) and α (right) cationic sites in the ferrierite framework. Silicon atoms are in gray, aluminum atoms are in yellow, and oxygen atoms are in red.
The α cationic site represents an elongated 6–membered ring which is composed of two 5–membered rings. This site is easily accessible because it is present on the wall of the main channel in ferrierite. The β cationic site in ferrierite corresponds to a deformed 6–membered ring. The β cationic site can be easily reached because it is positioned in an 8–membered ring channel of ferrierite.
4. Studied zeolites
We mainly studied the pentasil–ring Si–rich matrices4-5 (with Si/Al > ca. 8): ZSM–5 (MFI structure), TNU–9 (TUN structure), zeolite beta (*BEA structure), mordenite (MOR structure), and ferrierite (FER structure) and the SSZ–13 zeolite (it is not a pentasil–ring zeolite).
The ZSM–5 zeolite78 is the most important pentasil–ring zeolite. The protonic forms of ZSM–5 are used in fluid catalytic cracking (FCC).79 Fe–ZSM–5 is an important catalyst of oxidations of organic molecules.47-51, 80 Cu–ZSM–581 and Fe–ZSM–582 play an important role in SCR–NOx.
The TNU–9 zeolite is a more complex analog of the ZSM–5 zeolite.20, 83 TNU–9 has been investigated as a catalyst for both acid (e.g., the alkylation, isomerization, and disproportionation of hydrocarbons;84-91 and the methanol–to–hydrocarbons reaction (MTH)92) and redox reactions (selective catalytic reduction of NO over Cu–TNU–993 and Co–TNU–9,93 and, oxidation of propane over Cu–TNU–994).
Ferrierite zeolite95 is one of the industrialized types, which has excellent catalytic properties96 in skeletal isomerization of n–alkene,97 methanol to olefin,98 N2O decomposition,46 CO2 hydrogenation,99 dehydration of methanol and ethanol,100 and dimethyl ether carbonylation.101
Beta zeolites belong to the most widely used aluminosilicates applied as catalysts in petrochemical technologies,102 syntheses of chemical commodities,103 and in the catalytic conversion of biomass.61, 104-105 Co–beta zeolites are active in SCR–NOx.106
The protonic form of mordenite107 (i.e., H-mordenite) is an important industrial catalyst108 of the selective carbonylation of methanol and dimethyl ether with CO. Corma et al. showed that the catalytic properties of mordenite depended on the Al siting in the mordenite framework.109-110 Moreover, Cu–mordenites are promising materials to transform methane to methanol.111-113
SSZ–13 is a newer Si–rich zeolite of the chabazite structure with a high hydrothermal stability.114-115 Recent studies have shown that Cu loaded SSZ–13 zeolites with the chabazite (CHA) structure are very promising catalysts with an extreme potential in deNOx reactions.116-117 The SSZ–13 zeolite as a member of the family of small pore zeolites containing large cavities within their structure has been recognized as active and highly stable catalysts for the methanol–to–olefin (MTO) process.118-120
5. Computational methods and programs
Atomistic simulations of catalytic reactions over solid catalysts are a challenge to modern computational chemistry. To model a catalytic process, the computational method used should correctly evaluate: (a) the interactions between the reactants, transition states, intermediates, products on the one hand and the catalyst on the other hand; (b) the thermodynamics and kinetics of the catalytic reaction steps to provide reasonable values for reaction energies and barriers; (c) the method should also be able to correctly describe the structural complexity of the catalyst.
A twofold approach is needed to computationally model a catalyst. On the one hand, the theoretical model used must include all the important features (e.g., the active sites) as well as the structural complexity of the catalyst. On the other hand, the theoretical method employed has to be able to correctly describe all the important interactions. Since the computer time and resource demand grow polynomially with the size of the model, there is always a tradeoff between the size and complexity of the catalyst model on the one hand, and the theoretical method used on the other hand.
Reliable predictions of the NMR parameters (i.e., NMR shielding tensors,121 and moreover for quadrupolar nuclei, also nuclear quadrupolar coupling constants121 (CQ) and asymmetry parameters121 (η)) of zeolite framework atoms or extra–framework cations require both (i) accurate predictions of the local structure of the atoms of interest (e.g., framework AlO4- and SiO4 tetrahedra as well as extra–framework cations and their corresponding cationic sites) since the calculated NMR parameters are very sensitive to small changes of the local structure and (ii) reliable calculations of the corresponding NMR parameters from the optimized structures.
In order to realistically model the structure, reactivity, and properties of zeolites as well as to computationally investigate the catalytic activity of zeolite catalysts, a computational model of these structurally complex systems has to be built based on experimental structural data. The best structures are those obtained by X–ray crystallography and neutron diffraction crystallography at good resolution. The computational models have to include a large number of atoms.
5.1. Periodic DFT
Since zeolites are crystalline matrices, periodic DFT methods are well suited for their theoretical study. A development of these methods and advances in computer technology in the 2000s have allowed simulations based on the full crystalline lattice of zeolites employing pure DFT functionals.72, 122-126 Periodic DFT methods were extensively employed by the studies of this dissertation.3, 15, 17-18, 20-21, 27, 39-45, 54, 127-128 There is no need for artificial division of the studied system between the different layers (e.g. inner layer, outer layer) as is necessary when QM/MM methods are used.
Molecular dynamics (MD) based on periodic DFT emerged as a valuable means to simulate crystalline solid catalysts, allowing more complete sampling of the configuration space than only localizing minima on the potential energy surface. Our studies showed that using MD simulations, or other similar global optimization techniques which allowed the structural rearrangement of the cationic sites upon binding divalent cations, were needed to obtain reliable local structures of these cationic sites3, 15, 17-18, 20-21, 40-45 as well as vibrational frequencies15 of probe molecules coordinating to M(II). Moreover, we showed that MD computations permitted determining the Li+ and Na+ siting in ferrierite without having any experimental structural data regarding the siting of Li+ and Na+ cations available.17, 21
5.1.1. The VASP program
VASP,129-133 which was developed by Professor Hafner of the University of Vienna, Austria, is well suited for studying crystalline materials such as zeolites. VASP implements periodic DFT methods using plane–wave basis sets and pseudopotentials. For crystalline solids, VASP allows a relaxation (optimization) of the structure and the lattice parameters as well as molecular dynamics simulations. The advantages are as follows: using plane–wave basis sets and pseudopotentials allows feasible calculations of periodic systems containing hundreds of atoms in the unit cell.
Transition metal–exchanged forms of zeolites have been extensively computationally studied, however, the main drawback of the majority of the calculations in the past was the use of single determinant quantum chemistry methods (mainly DFT) which might fail in the case that the studied system is of multireference character (e.g., when the d–orbitals of the transition metal are partly occupied).134 Single reference methods are appropriate mainly for transition metals which have their d–orbitals either empty or fully occupied (e.g., Sc and Cu, respectively). Conversely, studies employing the VASP program have shown3, 15, 18, 20, 40-45, 54, 72, 125-128, 135-140 that periodic DFT methods employing plane–wave basis sets and pseudopotentials yield reasonable results for systems containing transition metals with partly occupied d orbitals. Such systems are well known to be difficult cases for single determinant quantum chemistry methods.134
5.1.2. The CP2K program
CP2K,141 which was developed by Professor Jürg Hutter of University of Zürich, Switzerland, is also an appropriate tool to study crystalline materials such as zeolites. CP2K features periodic DFT with several functionals and uses both plane waves as well as Gaussian functions. CP2K was employed for the optimizations of the structures regarding the Al siting in ferrite,27 the Li+ siting in ferrite,17 Na+ siting in ferrite,21 and the Al organization in the SSZ–13 zeolite.3
Our studies revealed that when periodic DFT was used to optimize the structure, and subsequently, clusters were cut out from the corresponding optimized super cells and used to compute the NMR parameters employing hybrid DFT functionals, the theoretical results were in very good accord with the observed values.3, 17, 21, 27
5.2. QM/MM
QM/MM methods represent a different approach than periodic DFT.142 The QM–Pot is a QM/MM approach developed for computationally studying zeolites.143-144 QM–Pot partitions the whole system (S) into two parts. The inner part (I) is treated by quantum mechanics (QM) and the outer part (O) as well as all the interactions between the inner and outer layers are treated by parametrized interatomic potential functions (Pot). The dangling bonds of the inner part are saturated by link hydrogen atoms. The atoms of the inner part together with the link atoms form the cluster (C).
Force fields are based on classical physics and they are heavily parameterized. Their applicability is limited to systems similar to those which were used to parameterize the force field. They cannot be used to study bond breaking/forming but can provide optimized geometries (structures) which are very close to the experimental ones.145 Sierka and Sauer developed a very successful force field (Pot) for zeolites used in the QM–Pot.145 We showed that the corresponding QM–Pot optimized clusters could be successfully used to calculate, utilizing the Gaussian program,146 the NMR parameters for atoms of interest employing the GIAO NMR method.147
5.2.1. The QMPOT program
The QM–Pot approach is implemented in the QMPOT program144 which utilizes the Turbomole program148-152 for the QM part and the GULP program153-154 for the periodic potential function calculations. QMPOT was employed for the optimizations of the structures concerning (i) the Al siting in ZSM–5,2, 26, 29 and furthermore, ZSM–22 and Theta–1 zeolites,28 (ii) Al Lewis sites in a zeolite of the chabazite structure,38 (iii) cationic sites in the dehydrated Li–, Na–, and K–forms of a zeolite of the chabazite structure,19 and (iv) the effects of Al/Si and Ge/Si substitutions and silanol nests on the local geometry of Al and Si framework sites and the 27Al and 29Si, respectively, NMR parameters in the SSZ–13 zeolite30 and the Ge and Al containing zeotype of the zeolite beta polymorph C (BEC) structure,155 respectively. The computed NMR parameters were in very good agreement with the experimental results. Our calculations employing hybrid DFT functionals19, 38, 155 provided somewhat better accord with the observed values than those using pure DFT functionals.2, 26, 28-30
5.3. Calculations of NMR parameters
The Gaussian program146 was used to compute the NMR parameters using the GIAO NMR method147 employing either the BLYP2, 26, 28-30 functional and TZVP2, 26, 28-30 basis set of Ahlrichs156 or the superior hybrid B3LYP3, 17, 19, 21, 27, 38, 155 functional and the pcS basis sets of Jensen:157 pcS–4 for the atom of interest and the cation if present in the computational model and pcS–1 for all the other atoms.3, 17, 19, 21, 27, 38, 155
5.4. Dispersion corrections
DFT methods improperly account for the important long–range London dispersion effects (van der Waals forces). Therefore, various dispersion correction schemes are used in our studies (the density–dependent energy correction (dDsC) dispersion correction158-159 for optimizations and the DFT–D2 method160 for molecular dynamics calculations).
6. Al organization: computational models and methods
The Al organization3 in the framework of Si–rich zeolites is a key property3-6, 20 and is defined in Table 1. The Al organization has been studied and defined only on zeolite matrices with the absence of Al–O–Si–O–Al sequences.3 Gaining insights into the Al organization3 is of utmost importance for development of highly selective catalysts.4-6, 8, 24-25
Table 1. Definition of terms regarding the Al organization3
|
Al siting7 |
Which different crystallographically distinguishable framework T sites are occupied by isolated single Al atoms. |
|
isolated single Al atoms |
These are unable to accommodate both bare Co(II) cations in dehydrated zeolites and Co(II) hexaaqua complexes in hydrated zeolites. |
|
Al pairs |
Two Al atoms separated by two or three Si atoms located in one 6–ring (only Al−O−(Si−O)2−Al) or 8–ring (both Al−O−(Si−O)2−Al and Al−O−(Si−O)3−Al) and forming a cationic site for bare divalent (Co(II)) cations in dehydrated zeolites. They are also able to accommodate Co(II) hexaaqua complexes in hydrated zeolites. |
|
close unpaired Al atoms |
Al atoms of Al−O−(Si−O)2−Al and Al−O−(Si−O)3−Al sequences accommodating Co(II) hexaaqua complexes in hydrated zeolites, but not bare Co(II) cations in dehydrated zeolites. These Al atoms have been observed only in beta zeolites18, 39 and SSZ–13.3 |
|
Al distribution |
The distribution of framework aluminum atoms among (i) Al−O−(Si−O)2−Al and Al−O−(Si−O)3−Al sequences in the rings forming cationic sites for bare divalent cations, (ii) Al−O−(Si−O)2−Al and Al−O−(Si−O)3−Al sequences corresponding to close unpaired Al atoms, and (iii) isolated single Al atoms. |
|
siting of Al pairs and close unpaired Al atoms |
Locations of Al atoms of Al−O−(Si−O)2−Al and Al−O−(Si−O)3−Al sequences corresponding to Al pairs and close unpaired Al atoms in the zeolite framework. |
|
siting of bare divalent cations |
Positions of bare extra–framework divalent cations in 6–rings and 8–rings of the zeolite framework. |
|
Al organization |
Al siting + Al distribution + siting of Al pairs, close unpaired Al atoms, and bare divalent cations + the location8 of isolated single Al atoms and Al−O−(Si−O)2−Al and Al−O−(Si−O)3−Al sequences of interest in the channel system of the zeolites that can be either in the channels or at the channel intersections. |
6.1. Development of the bare zeolite framework model – model of fully hydrated zeolites
High–resolution 27Al MAS NMR spectra of zeolites can be successfully measured only for fully hydrated matrices due to the strong quadrupolar interaction of the aluminum atoms in dehydrated zeolite.2, 19, 26-27, 29-30 Therefore, the NMR parameters characterizing the Al atoms and their environments in the zeolite frameworks can be measured only for zeolites containing counter cations or H3O+ and water molecules. Due to an enormous number of possible configurations, isotropic chemical shift calculations including the hydration of zeolite and the solvated counter cation would require extensive sampling over isotropic chemical shifts calculated quantum mechanically for the individual configurations and structures.2, 26-27, 29-30 To avoid this huge computational problem, we developed a simple model of the complex structure of a hydrated zeolite. We employ a bare charged framework with a single Al atom in a unit cell to describe the local geometry around the Al nucleus of isolated single Al atoms. Similarly, two Al atoms are used to characterize the local geometry about the two Al nuclei of Al−O−(Si−O)n−Al (n = 1, 2, and 3) sequences. Each Al atom bears a formal charge of -1. This is a realistic model because of the reasons as follows: in completely hydrated zeolites, the fully solvated counter cation is located close to the center of the cavity/channel and does not directly interact with the AlO4- tetrahedra as evidenced by a number of XRD studies of hydrated zeolites.161 Our study showed that the effect of the hydrated counter cation on the local geometry of the AlO4- tetrahedra in hydrated Si–rich zeolites and thus on the 27Al isotropic chemical shift was negligible.2, 26-27 The development of the bare framework model which includes neither water molecules nor explicitly counter cations represents the crucial step which permitted the computations of reliable local structures and NMR parameters for the zeolites of interest due to the simplification of the computational model. In addition, the bare framework model can be employed to calculate the local geometry of framework SiO4 tetrahedra and 29Si chemical shifts of framework Si atoms of both hydrated and dehydrated zeolites.30, 155 The bare framework model was used to determine the partial siting of isolated single Al atoms in a set of ZSM–5 zeolites,2, 26 Al siting in the ZSM–22 and Theta–1 zeolites,28 and the full siting of both isolated single Al atoms and Al–O–(Si–O)2–Al sequences in a set of ferrierite zeolites.27 Furthermore, this model was utilized to establish the effects of Al/Si and Ge/Si substitutions and silanol nests on the local geometry of Al and Si framework sites and the 27Al and 29Si, respectively, NMR parameters in the SSZ–13 zeolite30 and the Ge and Al containing zeotype of the zeolite beta polymorph C (BEC) structure,155 respectively. Moreover, the effects of the presence of Al–O–Si–O–Al and Al–O–(Si–O)2–Al sequences in the ZSM–5 zeolite framework on the local geometry of AlO4- tetrahedra and the 27Al NMR parameters were investigated employing the bare zeolite framework model.29 In addition, the same model was utilized to determine the siting of close unpaired Al atoms in the SSZ–13 zeolite.3
6.2. Models of dehydrated zeolites
Computational models of dehydrated zeolites are composed of framework atoms and extra–framework cations. The tetrahedral framework Al atoms are fully charge balanced. These models were utilized to investigate:
1) The quadrupolar interaction of 27Al nuclei in dehydrated zeolites.19
2) The Li+ and Na+ siting in Li–ferrierite17 and Na–ferrierite,21 respectively, matrices.
3) The structure and formation of Al Lewis sites.38-39
4) The structure and stability of cationic sites formed by bare divalent cations.3, 15, 18, 20, 40-45, 54, 127-128
6.3. Structure optimizations
There are two main approaches to optimization of the structure of zeolites in computational chemistry which are described in the next two Sections (6.3.1. and 6.3.2). The starting structures are generated from available structures determined by XRD or neutron diffraction.
6.3.1. The periodic DFT approach
6.3.1.1. The CP2K program
Both the lattice constants and the atomic positions of a super–cell of the zeolite of interest are optimized by the CP2K program.141
Structure optimizations using the bare framework model are employed to obtain the Al siting in ferrierite27 and to determine the siting of close unpaired Al atoms in SSZ–13.3 Conversely, the calculations to determine the Li+ siting in ferrite17 and Na+ siting in ferrite21 include molecular dynamics computations to properly sample all possible symmetrically non–equivalent Li+ and Na+ sites. Subsequently, the structures of distinct snapshots collected at regular intervals from the MD simulations are optimized for all the computational models. The most stable structures of all distinct cationic sites for all the models are used for subsequent NMR computations.
6.3.1.2. The VASP program
The atomic positions but not the lattice parameters of a super–cell of the zeolite of interest are optimized by the VASP program in the studies of the structure and stability of cationic sites formed by bare divalent cations3, 15, 18, 20, 40-45, 54, 127-128 in various zeolites, and furthermore, framework Al Lewis sites39 in the beta zeolite. These calculations also included MD simulations which permitted the structural rearrangement of (i) the cationic sites upon binding bare divalent cations and (ii) Al atoms tricoordinated to the zeolite framework. The structures of distinct snapshots collected at regular intervals from the MD simulations are optimized. The most stable structures are used for the studies.
6.3.2. The QM–Pot approach
Both the lattice constants and the atomic positions of the all–silica zeolite structure of interest (i.e., no Al atoms, only Si and O) are optimized at constant pressure by the GULP program153-154 using interatomic potential functions only.145 Then the Si atoms in the site of interest are replaced by an Al atom(s) and the structure and the lattice constants are further optimized at constant pressure. The optimized structures are subsequently used for defining a cluster around the Al (or Si) atom(s) of interest for QM–Pot.143-144 calculations. The clusters are embedded into a super cell composed of one or more–unit cells of the zeolite framework and the structure of the entire system is optimized by QMPOT144 at constant volume.
Clusters having the Al (or Si) atom of interest in the center and including five2, 19, 26, 28-30, 38, 155 coordination shells (i.e., Al–O–Si–O–Si–O–Hlink) are used. They are cut out from the corresponding optimized super cells. Due to the presence of silicate rings in the framework of zeolites, the created clusters contained pairs of very close Hlink atoms. Since the close Hlink atoms represented the same Si atom, they are replaced by the corresponding Si(OHlink)2 moiety. This is repeated until the cluster contained no such pairs. Double,29-30, 38, 155 triple,155 quadruple,155 and pentuple155 centered clusters can used in the same manner as the described single2, 19, 26, 28, 30 centered clusters.
6.4. Computation of the (isotropic) chemical shift
The cluster of five2, 19, 26, 28-30, 38, 155 coordination shells used in the QM–Pot optimizations or clusters of five27 and seven3, 17, 21 coordination shells around the atom of interest (i.e., Al−O−Si−O−Si−O−Hlink or Al−O−Si−O−Si−O−Si−O−Hlink) extracted from the periodic DFT optimized structures are utilized to calculate the 27Al NMR shielding using the GIAO NMR method.147 The counter–cations Li+,17, 19 Na+,19, 21 and K+19 cations are included in the cluster. Double,27, 29-30, 38, 155 triple,155 quadruple,155 and pentuple155 centered clusters can used in the same manner as the described single2, 19, 26, 28, 30 centered clusters.
Conversion of calculated NMR shielding values into the corresponding (isotropic) chemical shifts was carried out using either the primary or secondary standards. The calculated NMR shielding for [Al(H2O)6]3+ (primary standard) was used for the conversion in the study of the Al siting in ferrierite27 while the SSZ–13 zeolite30 with the Si/Al ratio of 38 was employed as a secondary standard in the studies of the Al siting in ZSM–5,2, 26 the ZSM–22 and Theta–1 zeolites,28 framework Al Lewis sites in the ferrierite and beta zeolites,38-39 and close unpaired Al atoms in SSZ–13.3 Similarly, [Li(H2O)4]+ and [Na(H2O)6]+ as the primary standards were used in the studies of the Li+ and Na+, respectively, siting in Li–ferrierite17 and Na–ferrierite,21 respectively. In addition, the conversion of 23Na NMR shielding into the 23Na isotropic chemical shifts for Na–ferrierite21 was further verified employing the Na–Y zeolite as a secondary standard. The difference between the assignments based on the primary and secondary standards was only 0.5 ppm, confirming a high reliability of the conversion of the calculated 23Na NMR shielding values to 23Na isotropic chemical shifts based on the primary standard.21
7. Al organization: results
7.1. Determination of the partial siting of isolated single Al atoms in ZSM–52, 26
We demonstrated investigating a set of eighteen differently synthesized ZSM–5 zeolites featuring predominantly isolated single Al atoms that a combined experimental (27Al (3Q) MAS NMR) and theoretical (QM–Pot employing the bare framework model) approach represented a powerful tool for the determination of the local geometry of framework AlO4- tetrahedra, the prediction of 27Al isotropic chemical shifts in hydrated zeolites, and the identification of Al siting in the framework of Si–rich zeolites. We determined that the occupation of the framework T–sites by Al and the concentration of Al in these T–sites were neither random nor controlled by a simple rule. They both depend on the conditions of the zeolite synthesis. Our study provided experimental evidence for the occupation of at least 12 out of 24 distinguishable framework T–sites by Al atoms in Si–rich ZSM–5. Figure 2 compares the calculated and measured 27Al isotropic chemical shifts.
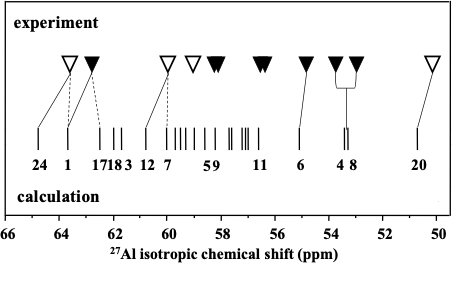
Figure 2. Comparison of the theoretical and observed 27Al isotropic chemical shifts in Na–ZSM–5 samples. Strong (▼) and weak (▽) resonances and Assignment 1 (- - -) and Assignment 2 (—) to the T1 – T24 sites. Adapted Figure 5 from Ref.26
The values for the different sites extend over similar ranges: 14.1 (calculated) and 13.6 (observed) ppm. In addition, the patterns of the predicted and observed isotropic chemical shifts show similarities. Based on similar spacings and least deviations between calculated and observed isotropic chemical shifts two partial assignments (Figure 2) of the observed resonances to the crystallographic sites in ZSM–5 are possible. To which of the remaining 13 T sites the unassigned resonances belong remains open. The results represent significant progress in interpretating 27Al MAS NMR spectra of zeolites and the first partially successful analysis of the Al siting in a framework of a Si–rich zeolite. Our study allowed only a partial identification of the Al siting because the ZSM–5 zeolite features 24 distinct AlO4- tetrahedra corresponding to 24 values of the 27Al isotropic chemical shifts. However, the NMR parameters of some T sites are too close to be distinguished by 27Al (3Q) MAS NMR spectroscopy. Additional methods are required, for example monovalent cations Li+17 and Na+21 as probes in dehydrated zeolites monitored by 7Li and 23Na (3Q), respectively, MAS NMR spectroscopies.
Our results also show that although there is a trend for smaller 27Al isotropic chemical shifts with increasing average Al–O–Si bond angles, the correlation is not good enough for assignment purposes. Therefore, the local geometry of framework AlO4- tetrahedra cannot be inferred from experimental isotropic chemical shifts but can only be obtained from theoretical calculations.
7.2. Effect of Al–O–Si–O–Al and Al–O–(Si–O)2–Al pairs in the ZSM–5 zeolite framework on the 27Al NMR spectra29
The effect of the presence of Al–O–Si–O–Al and Al–O–(Si–O)2–Al sequences in the ZSM–5 zeolite framework on the local geometry of AlO4- tetrahedra and the 27Al NMR parameters was investigated employing 27Al 3Q MAS NMR spectroscopy and QM–Pot calculations. The Al–O–(Si–O)2–Al chains form the a and b cationic sites for bare divalent cations. Our calculations revealed that the presence of an Al atom as a next–nearest (Al–O–Si–O–Al) and next–next–nearest (Al–O–(Si–O)2–Al) neighbor could significantly affect both the local geometry of AlO4- tetrahedra as well as 27Al NMR isotropic chemical shift (up to 4 ppm), see Figures 3 and 4.
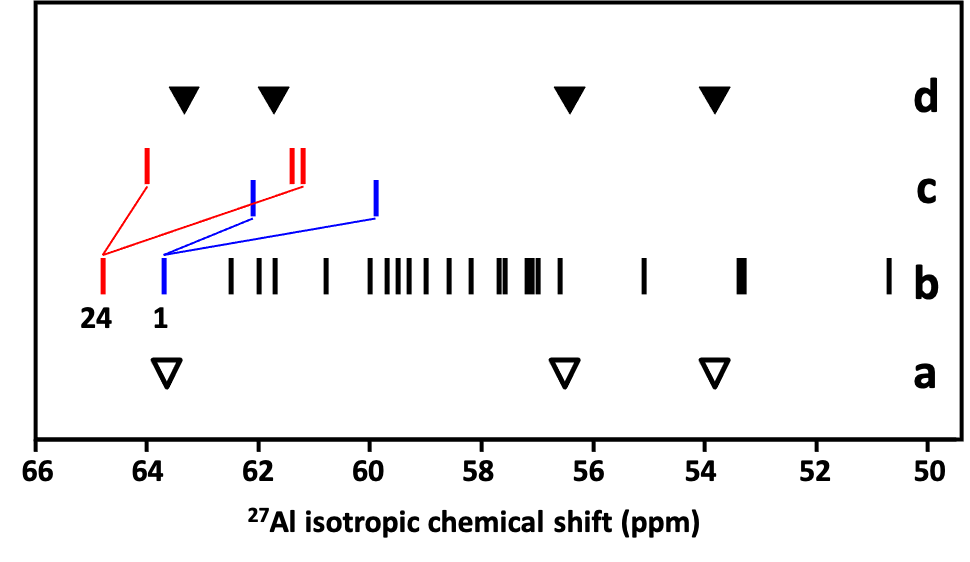
Figure 3. Effect of the presence of an Al atom as a next–nearest neighbor on the calculated 27Al isotropic chemical shift of the Al atoms occupying T1 and T24 sites (Al(T1) and Al(T24), respectively). Experimental values of 27Al isotropic chemical shift for the ZSM–5 sample with no Al–O–Si–O–Al sequences (a), calculated isotropic chemical shifts of isolated single Al atoms in ZSM–52 (b), calculated isotropic chemical shifts of Al(T1) (blue) and Al(T24) (red) present in Al–O–Si–O–Al sequences (c), and experimental values of 27Al isotropic chemical shift for the ZSM–5 sample with Al atoms located predominantly in Al–O–Si–O–Al sequences (d). (▼) Al possibly in pairs; (▽) possible single Al. Adapted Figure 10 from Ref.29
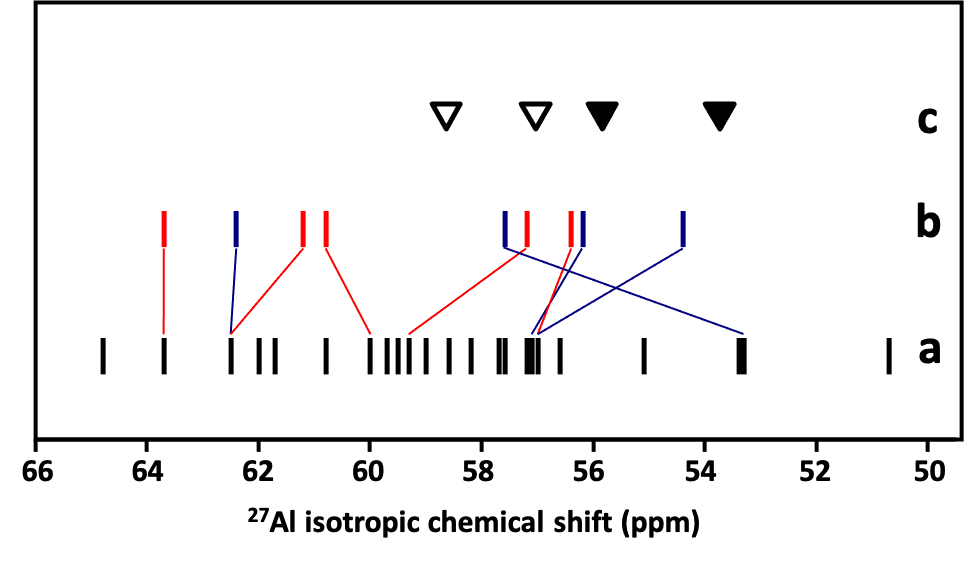
Figure 4. Effect of the presence of an Al atom as a next–next–nearest neighbor on the calculated 27Al isotropic chemical shift of the Al atoms of the α and β cationic sites. Calculated isotropic chemical shifts of isolated single Al atoms in ZSM–52 (a), calculated isotropic chemical shifts of Al present in the α (blue) and β (red) cationic sites (b), and experimental values of 27Al isotropic chemical shift of the two ZSM–5 samples with Al atoms located predominantly in Al–O–(Si–O)2–Al sequences (c). (▼) Al possibly in pairs; (▽) possible single Al. Adapted Figure 11 from Ref.29
There is no systematic contribution of Al in Al–O–Si–O–Al or Al–O–(Si–O)2–Al chains to the 27Al isotropic chemical shift, and not even the direction can be predicted without explicit DFT calculations. Our investigation showed that the method to determine the Al siting in ZSM–5 used in our study2, 26 can be employed only for ZSM–5 samples having a low or negligible concentration of Al–O–Si–O–Al and Al–O–(Si–O)2–Al sequences in the zeolite matrix, otherwise 27Al (3Q) MAS NMR spectroscopy cannot be used to even identify the number of framework T sites occupied by Al.
7.3. The location of isolated single Al atoms and Al−O−(Si−O)2−Al sequences of interest in the channel system of the zeolites8
The location can be either in the channels or at the channel intersections. Our study on ZSM–5 zeolites shows that zeolites prepared using exclusively TPA+ as a structure–directing agent (i.e., in the absence of Na+ cations) led to 55–90% of Al atoms located at the channel intersection, regardless the presence or absence of Al pairs in the zeolite framework. The presence of Na+ cations in the synthesis gel did not modify the Al location at the channel intersection (55–95% of Al atoms) and led only to changes in i) the distribution of framework Al atoms between Al pairs (decrease) and single isolated Al atoms (increase), and ii) the siting of Al in distinguishable framework tetrahedral sites.
7.4. Determination of the siting of isolated single Al atoms in ZSM–22 and Theta28
Our QM/MM calculations in tandem with the already published 27Al 3Q MAS NMR experimental data on the Si–rich ZSM–22 and Theta–1 zeolites of the TON structure showed that Al atoms could be located in 6 framework T positions because the two eightfold sites (T1 and T2) split into four fourfold T sites after an Al/Si substitution (Figure 5).
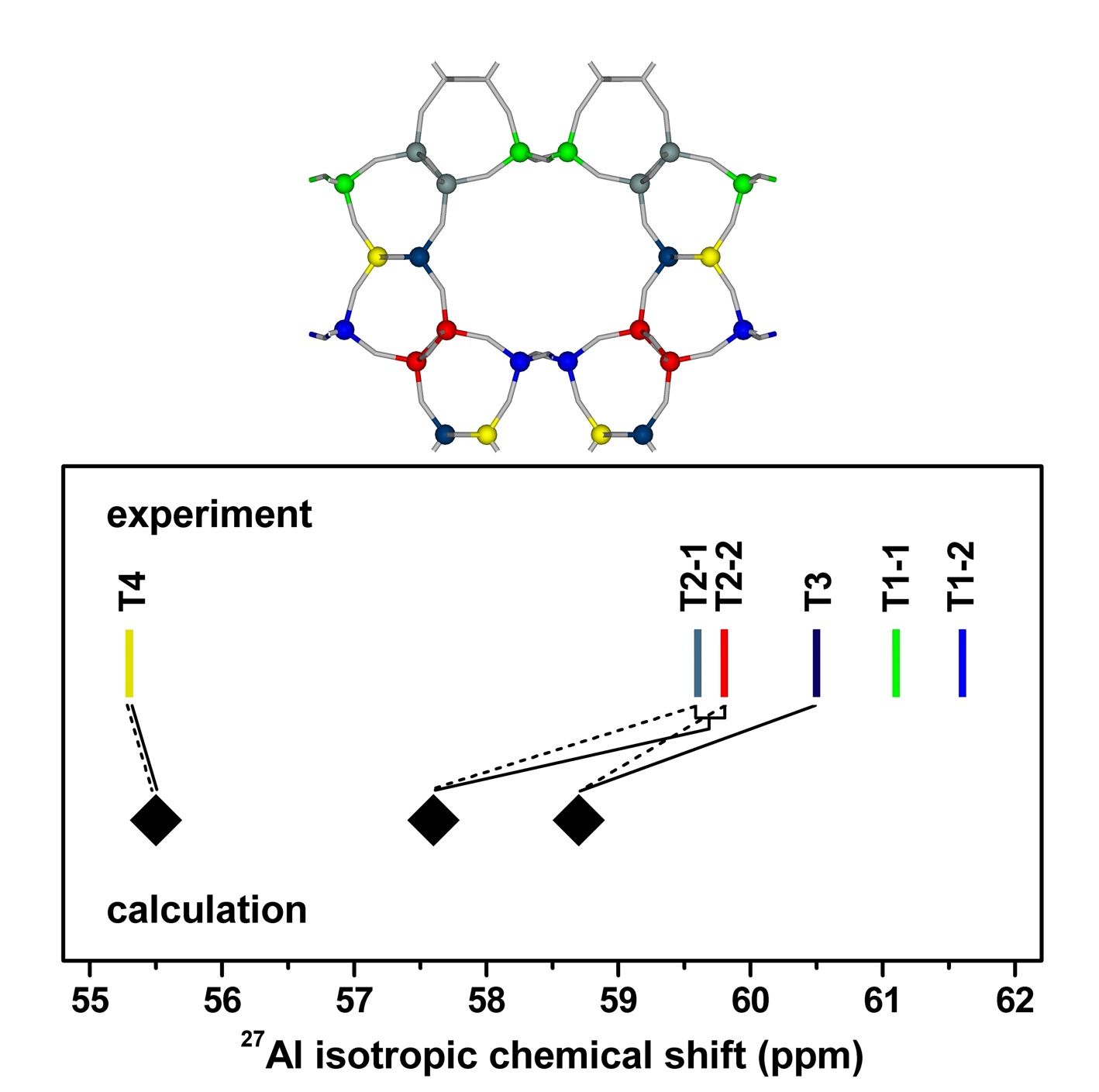
Figure 5. Unit cell with showing the calculated T sites. Comparison of the observed and theoretical 27Al isotropic chemical shifts of Theta–1; Assignment 1 (—) and Assignment 2 (- - -). Adapted Figures 1 and 3 from Ref.28
Al atoms in the T4 site predominate in the Theta–1 and ZSM–22 zeolites (about 60%). Some 40% of Al atoms are located in the T2 site, and the T3 site accommodates only a minority of Al atoms (10%) or is together with the T1 site unoccupied by Al atoms. The T4 site is not located on the surface of the TON 10–ring channel and thus the protonic sites related with the majority of Al atoms in the TON structure exhibit a significantly limited reaction space.
7.5. Complete determination of the siting of Al atoms in Si–rich zeolites of the ferrierite structure27
Employing a set of five differently synthesized ferrierite zeolites we developed a multistep method allowing determination of the complete Al distribution in Si–rich zeolites with fewer crystallographically distinguishable framework T sites independent of the presence of Al–O–Si–O–Al or Al–O–(Si–O)2–Al sequences in their frameworks. This approach combined multispectroscopic experiments with periodic DFT calculations.
The complete Al siting in the three ferrierite samples with only isolated single Al atoms and two ferrierites with Al–O–(Si–O)2–Al sequences was determined. Our results (Figure 6) reveal that the Al siting in the samples with only isolated single Al atoms (A, B, and C) varies with the conditions of the zeolite synthesis.
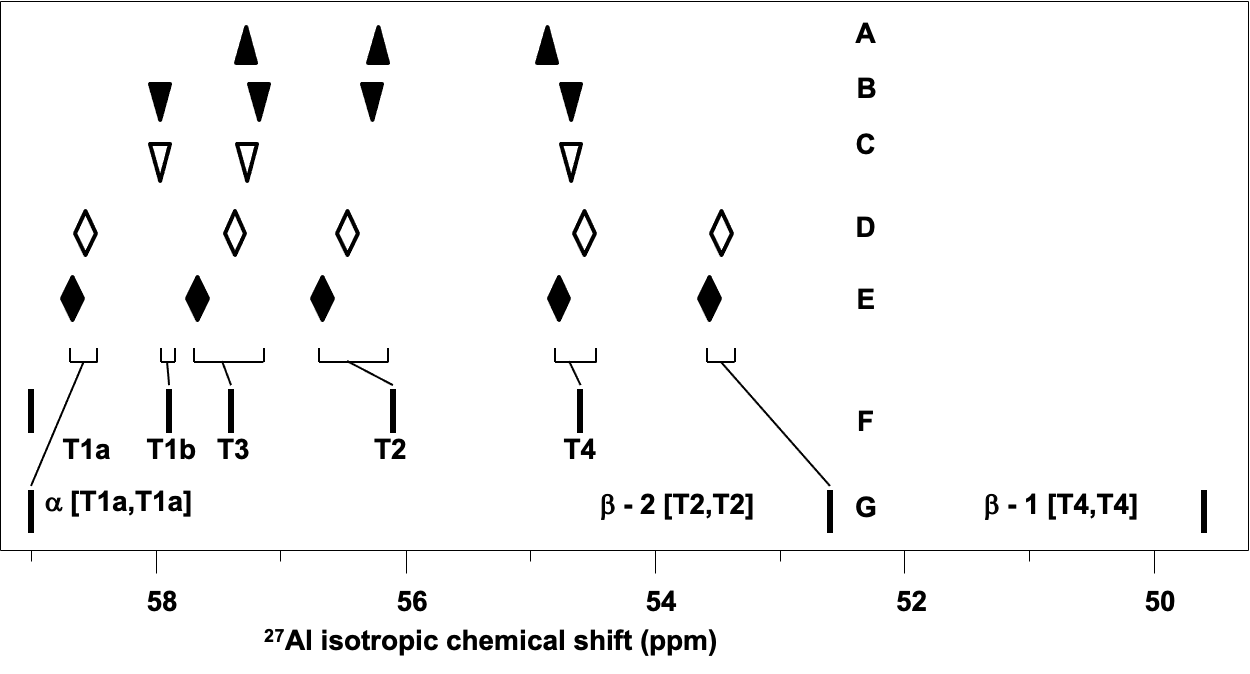
Figure 6. Comparison of the observed (A–E) and calculated (F–G) 27Al isotropic chemical shifts for ferrierites shows the assignment of the 27Al NMR resonances to the T sites. The experimental data for the ferrierites (i) with predominantly isolated single Al: (A) FER/A, (B) FER/B, (C) FER/C and (ii) with isolated single Al as well as Al–O–(Si–O)2–Al sequences: (D) FER/D, (E) FER/E. 27Al isotropic chemical shifts calculated for isolated single Al atoms (F), and for Al–O–(Si–O)2–Al sequences in the α and β cationic sites (G). Adapted Figure 5 from Ref.27
The 16–fold T1 site splits into two 8–fold subsets (T1a and T1b) of the T1 site. Al is present in three or four sites (T1b, T2, T3, and T4) depending on the samples with only isolated single Al atoms (A, B, and C) while T1a is never occupied by Al and the concentrations of Al atoms in various T sites are very diverse. For the two ferrierites with both isolated single and close Al atoms (D and E), isolated single Al atoms occupy the T2, T3, and T4 sites and the Al pair atoms are arranged in Al(T1a)–O–(Si–O)2–Al(T1a) and Al(T2)–O–(Si–O)2–Al(T2) sequences forming the α and β–2 cationic sites, respectively. Isolated single Al atoms do not occupy the T1b site and close Al atoms do not form Al(T4)–O–(Si–O)2–Al(T4) sequences of the β–1 cationic sites in the D and E samples. The differences between the concentrations of Al in T sites are not as pronounced as those for the ferrierite samples with only isolated single Al atoms. In addition, our results reveal that the Al siting in ferrierite is not random and depends on the conditions of the zeolite synthesis.
7.6. Effect of Al/Si substitutions and silanol nests on the local geometry of Si and Al framework sites in Si–rich zeolites30
29Si and 27Al (3Q) MAS NMR spectroscopy and QM–Pot calculations were employed to investigate the effect of Al/Si substitutions and the presence of silanol nests on the 29Si and 27Al NMR parameters as well as the local geometry of SiO4 and AlO4- tetrahedra of the nearest and next–nearest neighboring Si and Al atoms. The Si–rich zeolite of the chabazite structure (Si/Al 38) was chosen for this study as a representative model of Si–rich zeolites since it exhibits a low number of distinguishable T sites. Our computational results show the following: (i) Al atoms can occupy three different crystallographic T sites in the framework of chabazite (Si/Al 38). This result is in agreement with two observed 27Al NMR resonances. (ii) An Al/Si substitution causes a downshift of the 29Si chemical shift of the nearest neighboring Si atoms (Al–O–Si) by 4–11 ppm. (iii) The effect of a more distant Al/Si substitution (Al–O–Si–O–Si) is significantly less pronounced (downshift up to 2 ppm). (iv) An Al/Si substitution (Al–O–Si–O–Al) leads to larger 27Al isotropic chemical shifts of the next–nearest neighboring Al atoms by up to 3 ppm. (v) The presence of a silanol “nest” (vacant T site) as a nearest (H–O–Si) and next–nearest (H–O–Si–O–Si) neighbor is responsible for a systematic downshift of the 29Si chemical shift of Si by 11–16 ppm and by 0–1 ppm, respectively. (vi) There is no systematic effect of a silanol “nest” as a next–nearest neighbor (H–O–Si–O–Al) on the 27Al isotropic chemical shift of Al as its values are smaller for some H–O–Si–O–Al sequences (up to -3.6 ppm) and greater for others (up to +2.9 ppm). (vii) Al atoms present in hypothetical Al–O–Al sequences would have their 27Al isotropic chemical shifts larger by 7–9 ppm than single Al atoms.
7.7. Formation and local structure of framework Al Lewis sites38-39
Employing high resolution 27Al MAS NMR and QM–Pot calculations we show38 that Al framework (AlFR) Lewis sites formed as minor species created under 300 °C in a zeolite of the FER structure are formed by dehydroxylation of terminal –(SiO)3–AlOH entities tricoordinated to the zeolite framework. The AlFR Lewis sites are reflected in an extremely broad 27Al NMR resonance with δiso ≈ 67 ppm and CQ ≈ 20 MHz. Such terminal AlFR Lewis sites are located at internal or external surfaces and are accessible to probe molecules and reactants. We conclude39 that the observed extremely broad 27Al NMR resonance corresponds to AlFR Lewis sites tricoordinated to the zeolite framework with adsorbed H2O. Our calculations yielded δiso = 59 ppm and CQ = 16.7 MHz for this site. These theoretical values are in good agreement with the experiment.
Framework AlFR Lewis sites represent a substantial portion of active sites in beta zeolite catalysts activated at low temperatures. We study39 their nature by 27Al WURST–QCPMG nuclear magnetic resonance (NMR) and propose a plausible mechanism of their formation based on periodic DFT calculations constrained by 1H MAS, 27Al WURST–QCPMG, and 29Si MAS NMR experiments and FTIR measurements. We suggest that these AlFR Lewis sites are formed from Al–OH–Si–O–Si–O–Si–OH–Al sequences located in 12–rings (i.e., close unpaired Al atoms). Our results show that the electron–pair acceptor of AlFR Lewis sites corresponds to an AlTRI atom tricoordinated to the zeolite framework, which adsorbs a water molecule (Figure 7).
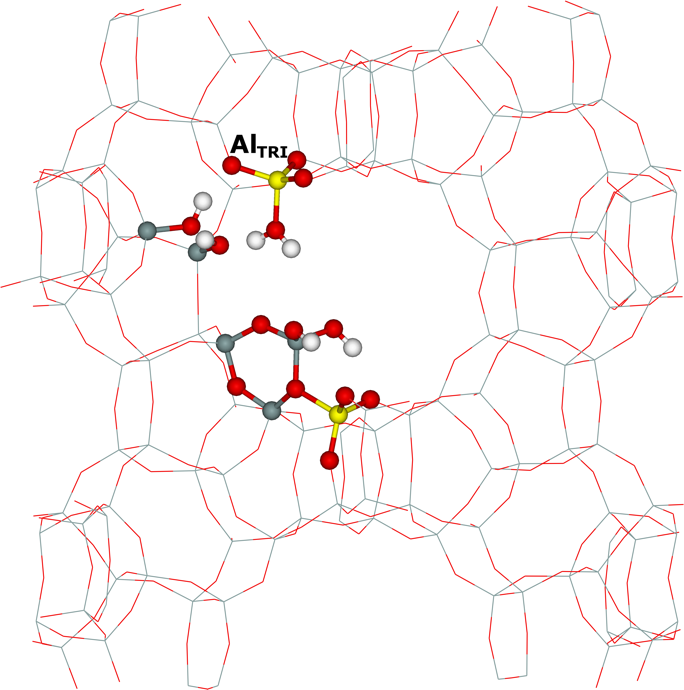
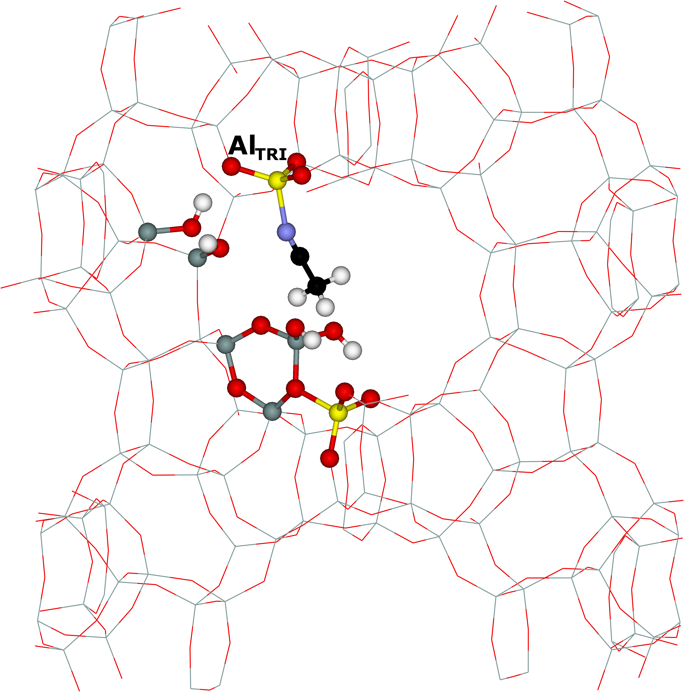
Figure 7. The optimized structures of the suggested model of the AlFR Lewis site in the beta zeolite with water (left) and acetonitrile (right) adsorbed on the AlTRI atom. The atoms of interest are displayed as balls. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, nitrogen atoms in blue, carbon atoms in black, and hydrogen atoms in white. Adapted Figure 6 from Ref.39
This AlTRI–OH2 complex is reflected in 27Al NMR resonance with δiso = 70 ± 5 ppm and CQ = 13 ± 2 MHz. In addition, the AlTRI atom with adsorbed acetonitrile–d3 (the probe of AlFR Lewis sites in FTIR spectroscopy) exhibits a similar 27Al NMR resonance (Figure 7). The latter finding was subsequently confirmed by Copéret et al. in the mordenite zeolite.162
7.8. Siting of Li+ cations as probes in dehydrated zeolites monitored by 7Li MAS NMR spectroscopy17
We developed a new method to determine the siting of Li+ and the local structure of Li+ sites
in crystalline aluminosilicate matrices based on a combination of 7Li–7Li correlation MAS NMR spectroscopy and periodic DFT calculations of the structure of Li+ sites and subsequent DFT cluster computations of the 7Li NMR shielding. The developed approach can be in general applied to Li+ cations in other zeolites and various crystalline matrices with large unit cells and a low concentration of Li+ cations without a significant limitation of their concentration. Our study shows that calculations with an extensive conformational sampling of the cation are required (due to the absence of experimental data regarding the siting of the cation) to obtain the accurate siting of the cation, i.e., employing only optimizations of the structure of the cationic sites in the zeolite framework is not sufficient.
Our study of the ferrierite zeolites with isolated single Al atoms (the same three samples as in our prior 27Al (3Q) MAS NMR study27 were used) reveals that Li+ cations occupy six distinct cationic sites. Two Li+ sites are occupied concurrently for Al(T1) and Al(T2) while only one for Al(T3) and Al(T4). Figure 8 shows the optimized structures of the calculated cationic sites.
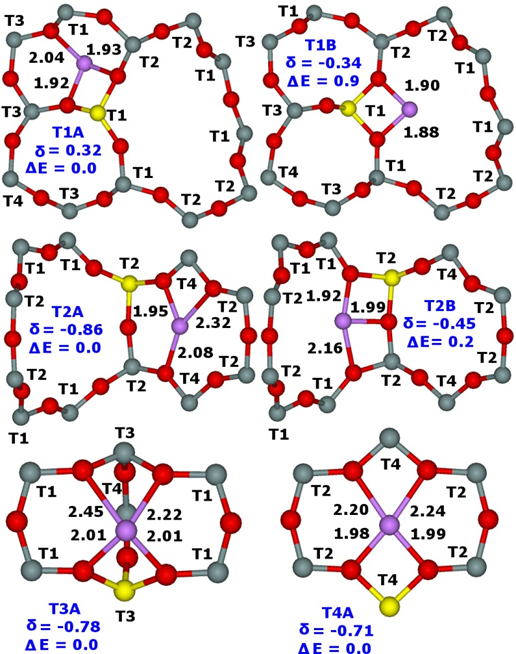
Figure 8. Optimized structures (Li–O distances in Å) with the designations of the T sites of the low energy Li+ sites (T1A and T1B for Al(T1), T2A and T2B for Al(T2), T3A for Al(T3), and T4A for Al(T4)), the relative energies in kcal/mol, and the corresponding 7Li chemical shifts in ppm converted using the 7Li NMR shielding of 90.16 ppm for Li+(H2O)4 with the 7Li chemical shift at 0.00 ppm. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and lithium in violet. Adapted Figure 3 from Ref.17
Figure 9 compares the experimental 7Li chemical shifts with the calculated ones for the three ferrierite samples already used in our determination of the siting of Al atoms in Si–rich zeolites of the ferrierite structure.27
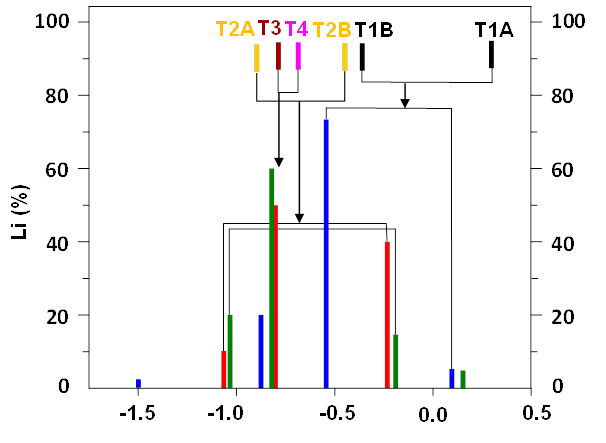
Figure 9. Experimental 7Li chemical shifts and their intensities in the spectra of the three dehydrated samples, 7Li chemical shifts calculated for Li+ cations balancing Al atoms in the T1 – T4 sites, and their assignments to the experimental data. The ferrierite samples with Si/Al 20 ( ▌), 27 ( ▌), and 30 ( ▌); Li+ balancing Al in the T1 ( ▌), T2 ( ▌), T3 ( ▌), and T4( ▌) sites. Adapted Figure 2 from Ref.17
7.9. Siting of Na+ cations as probes in dehydrated zeolites monitored by 23Na (3Q) MAS NMR spectroscopy21
We developed a method for the analysis of the siting of monovalent Na+ cations in extra–framework cationic sites in Si–rich ferrierite zeolites. The Na+ siting was analyzed by a combination of high–field (500 MHz) and ultra–high–field (900 MHz) MAS and MQMAS 23Na NMR spectroscopy interpreted using predictions of the 23Na NMR parameters (i.e., 23Na NMR shielding tensor, nuclear quadrupolar coupling constants (CQ), and asymmetry parameters (h)) obtained from periodic DFT calculations including extensive MD simulations. The employment of high–field and ultra–high–field 23Na solid–state NMR allows estimations of the 23Na NMR parameters of Na+ cations in different cationic sites with a high reliability. On the one hand, 23Na MAS NMR spectroscopy is essential for the identification of Na+ cations with a high quadrupolar broadening. 23Na NMR resonances of such cations are suppressed in 23Na MQMAS NMR experiments. On the other hand, 23Na MQMAS NMR measurements are essential for the analysis of close overlapping resonances with a lower quadrupolar broadening.
Our study of the ferrierite zeolites with isolated single Al atoms (the same three samples as in our prior 27Al (3Q) MAS NMR study27 were used) shows that Na+ cations can occupy nine distinct extra–framework cationic sites created by two 6–rings and two 8–rings with one Al atom located in different framework T sites. Eight cationic sites are occupied by Na+ in the three ferrierite samples used. 5–rings do not form cationic sites for Na+ cations.
The relationship between the calculated and observed 23Na isotropic chemical shifts δiso and the calculated and observed nuclear quadrupolar coupling product PQ (PQ = CQ(1 + η2/3)1/2) of Na+ cations is compared in Figure 10.
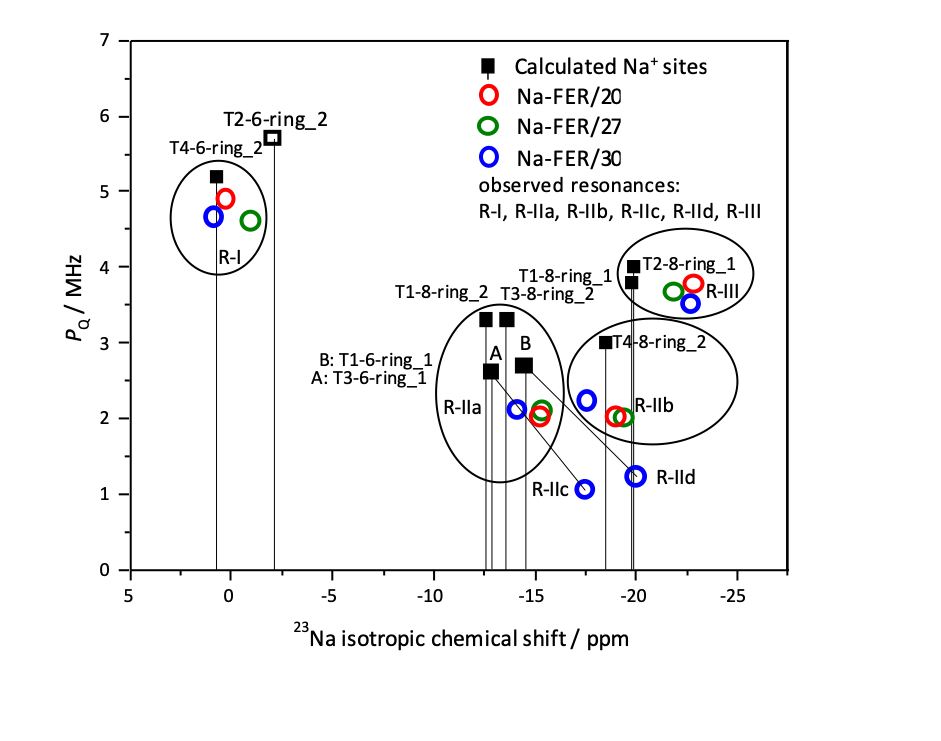
Figure 10. Comparison of the relationship between the calculated (squares) and observed (circles) nuclear quadrupolar coupling product (PQ) and the calculated (squares) and observed (circles) 23Na isotropic chemical shifts for the nine calculated Na+ cationic sites. The black squares correspond to the cationic sites occupied by Na+ while the white square relates to the most likely unoccupied cationic site. Adapted Figure 11 from Ref.21
The Al siting in the rings forming the Na+ cationic sites is not markedly reflected in the Na+ coordination and thus in the corresponding 23Na NMR parameters. Therefore, 23Na solid–state NMR spectroscopy can clearly identify the ring accommodating the Na+ cation, while the Al siting in the ring can be determined only for special cases.
Our study shows that 23Na solid–state NMR spectroscopy represents, on the one hand, a highly promising tool for the analysis of the siting of monovalent cations in Si–rich zeolite matrices, and, on the other hand, a method permitting the identification of the rings with an Al atom, and when applied together with 27Al and eventually 7Li solid–state NMR spectroscopies, it may allow the full analysis of the Al siting in Si–rich zeolites with a high number of framework T sites.
7.10. Investigation of the quadrupolar interaction of 27Al nuclei in dehydrated zeolites19
We studied the quadrupolar interaction of 27Al nuclei in dehydrated M–forms (M = Li, Na, and K) of chabazite using high–resolution 27Al MAS NMR spectroscopy together with DFT calculations to understand the corresponding high–resolution 27Al MAS NMR spectra. We show that the broadening of the 27Al NMR signal in dehydrated zeolites occurs predominantly because of the deformation of the local structure of AlO4- tetrahedra caused by the binding of M+ to the zeolite framework. This deformation increases with the decreasing diameter of the cations from K+ to Li+. The influence of water in hydrated zeolites is limited only to prevent a strong coordination of the M+ cation to O atoms of the AlO4- tetrahedron, but there is no “averaging” effect concerning the local electrostatic field due to molecular motion of water molecules. Our results show that the 27Al NMR parameters in dehydrated zeolites can be calculated accurately enough to permit the description of the local structure of AlO4- tetrahedra in dehydrated zeolites and to infer the local structure of the sites accommodating the extra–framework M+ cations.
7.11. Effects of single and multiple Ge/Si substitutions on the 29Si NMR parameters and the local geometry of SiO4 tetrahedra of the nearest (Ge–O–Si) and next–nearest (Ge–O–Si–O–Si) neighboring Si atoms in zeolites155
Employing the zeolite Beta polymorph C (BEC) we examined the effects of one, two, three, and four framework T (T = Ge and Al) atoms as the nearest (T–O–Si) neighbors on the 29Si chemical shift and the SiO4 local geometry. Our calculations reveal a systematic downshift of the 29Si chemical shift of Si by 1–6 ppm and 3–11 ppm for Ge–O–Si and Al–O–Si sequences, respectively. Furthermore, our results show that the contributions of two, three, and four Ge atoms as the nearest neighbors to the downshift of Si are not additive and the calculated downshifts lie in the intervals from 2 to 6 ppm, from 1 to 9 ppm, and from 5 to 11 ppm, respectively. Conversely, the contributions of two, three, and four Al atoms as the nearest neighbors are approximately additive and the calculated downshifts range from 7 to 18 ppm, from 14 to 23 ppm, and from 20 to 25 ppm, respectively.
Our calculations show that there is no systematic contribution of T (T = Ge and Al) atoms as next–nearest (T–O–Si–O–Si) neighbors to the 29Si chemical shift of Si, and not even the direction (sign) can be predicted without calculating the corresponding sequence. The effect is ± 1 and ± 2 ppm for the majority of the Ge and Al atoms, respectively.
7.12. Structure of Fe(II),40, 127 Co(II),15 and Cu(II)15 cationic sites in ferrierite
Accommodation of Fe(II) cations in the α and β cationic sites of the ferrierite zeolite were investigated using periodic DFT calculations firstly without employing molecular dynamics simulations127 but subsequently we found that the inclusion of molecular dynamics simulations was required because the binding of bare divalent cations to oxygen atoms of the rings forming cationic sites can lead to significant rearrangements of the local structures of the zeolite framework.40
The structure of sites binding divalent cations were estimated using X–ray diffraction experiments,75-77, 163-164 however, the obtained structures of the cationic positions represented a superposition of different arrangements of these sites with and without accommodated divalent cations. Moreover, some rings forming vacant cationic sites contain two Al atoms which are needed for the site to ligate a bare divalent cation, while other rings include only one or no Al atom (then the rings are not able to accommodate a bare divalent cation). These issues result in a significant decrease in the accuracy of the X–ray diffraction determined structure of the framework oxygen atoms involved in coordinating the divalent cation.40 Conversely, the structure of sites accommodating a divalent cation can be inferred from theoretical calculations.40
The starting structures used were generated from the experimental orthorhombic structure of ferrierite determined by neutron diffraction.165 Divalent metal cations located in sites prefer to coordinate to oxygen atoms of AlO4- tetrahedra rather than SiO4 tetrahedra, since the cations compensate the negative charge of AlO4-. However, the starting structures featured this proper coordination of Fe(II) to four oxygen atoms of two AlO4- tetrahedra only for the β–1 cationic sites formed by Al(T4)–O–(Si–O)2–Al(T4) sequences. Conversely, the Fe(II) cation is ligated to only three and two oxygen atoms of two AlO4- tetrahedra for the α and β–2, respectively, cationic sites created by Al(T1a27)–O–(Si–O)2–Al(T1a27) and Al(T2)–O–(Si–O)2–Al(T2), respectively, chains. Therefore, the structure of the α and β–2 cationic sites accommodating Fe(II) significantly rearranged during the MD calculations while that of the β–1 cationic site did not. The Fe(II) cation correctly coordinates to four oxygen atoms of two AlO4- tetrahedra in the resulting structures (Figures 11 and 12) for all the three cationic sites.
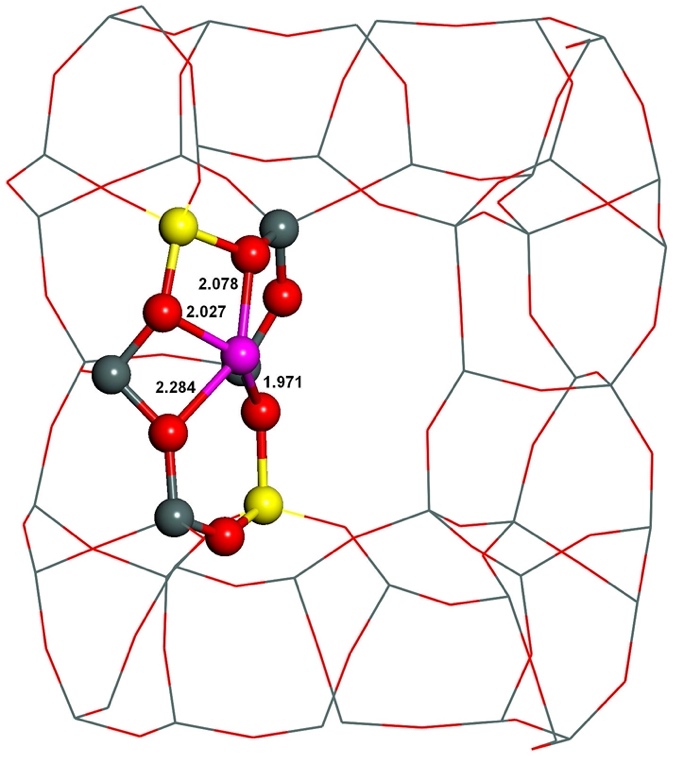
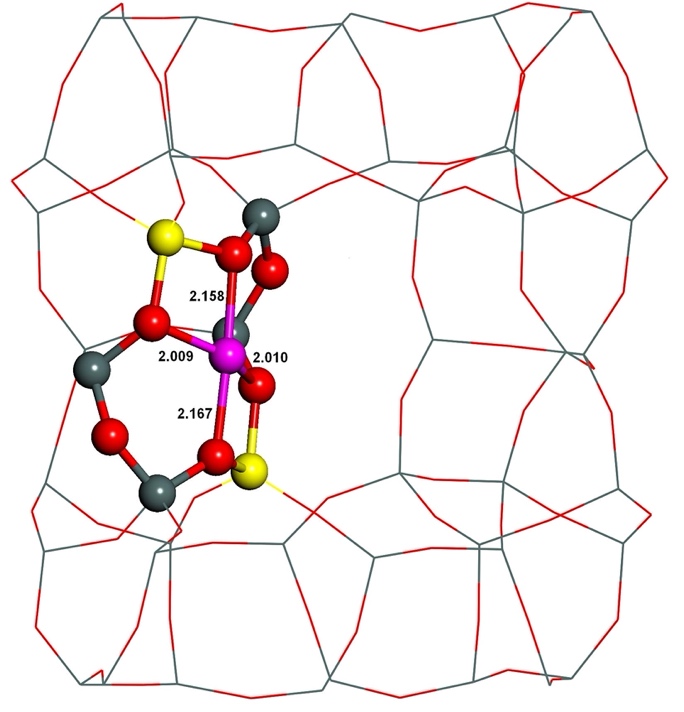
Figure 11. Optimized structures of the α cationic site before (left) and after (right) molecular dynamics simulations. The distances are in Å. Silicon atoms are in gray, aluminum atoms in yellow, iron atoms in violet, and oxygen atoms in red. Adapted Figure 7 from Ref.40
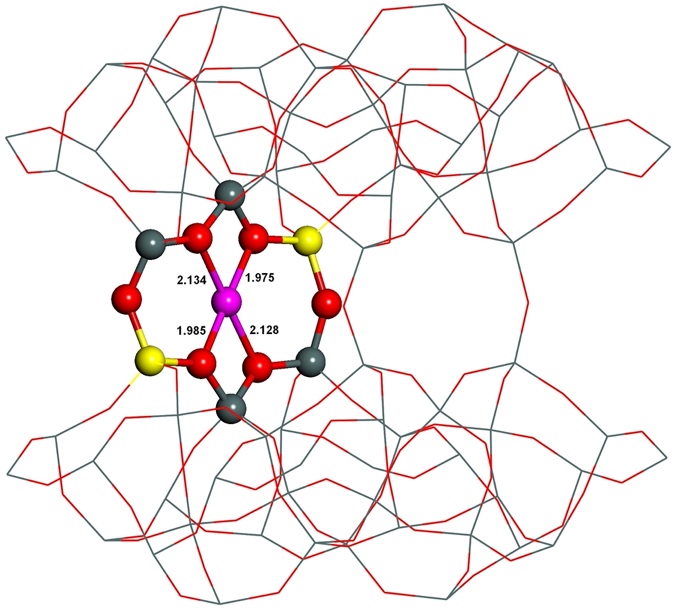
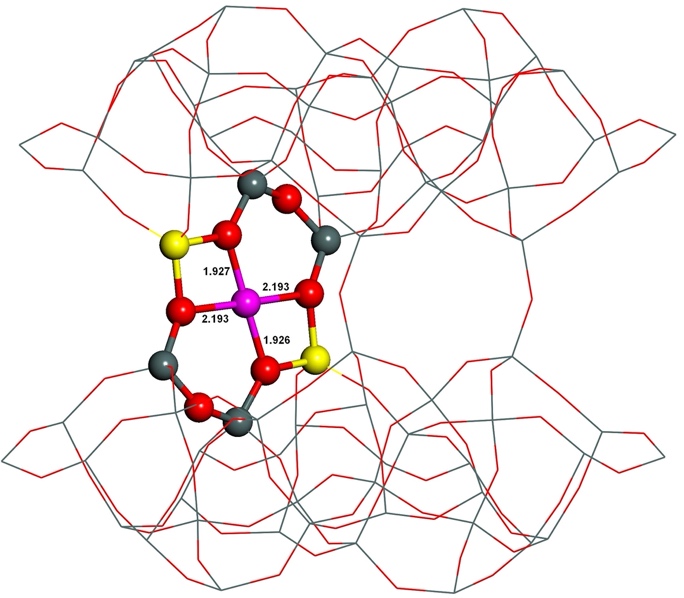
Figure 12. Optimized structures of the β–2 cationic site before (left) and after (right) molecular dynamics simulations. The distances are in Å. Silicon atoms are in gray, aluminum atoms in yellow, iron atoms in violet, and oxygen atoms in red. Adapted Figure 9 from Ref.40
The stabilization of the α and β–2 cationic sites due to the rearrangement is calculated to be about -11 to -12 kcal/mol per site while the relaxation of the framework for the β–1 cationic site leads to a small drop of the energy.
Very similar results were obtained for the accommodation of Cu(II) and Co(II) cations in the α and β cationic sites of the ferrierite zeolite.15 The α and β–2 cationic sites markedly rearranged upon Cu(II) and Co(II) binding to reach the proper coordination of Cu(II) and Co(II) to four oxygen atoms of two AlO4- tetrahedra.15
Furthermore, the interaction of Cu(II) and Co(II) ligated in the three cation sites (i.e., α, –1, and β–2) with a probe molecule (NO) was investigated employing FTIR spectroscopy and periodic DFT calculations including extensive molecular dynamics computations to study the local coordination environment of the Cu(II) and Co(II) cations located in the active sites and to validate the calculated results.15 Our calculations allow assignments of the experimental IR bands to the NO–M–ferrierite complexes: 1956, 1941, and 1935 cm−1 to NO–Co located in the α, β–1, and β–2 sites, respectively; 1864, 1912, 1904, and 1892 cm−1 to NO–Cu accommodated in the α, β–1, β–2 (conformer 1), and β–2 (conformer 2) sites, respectively (Figure 13).
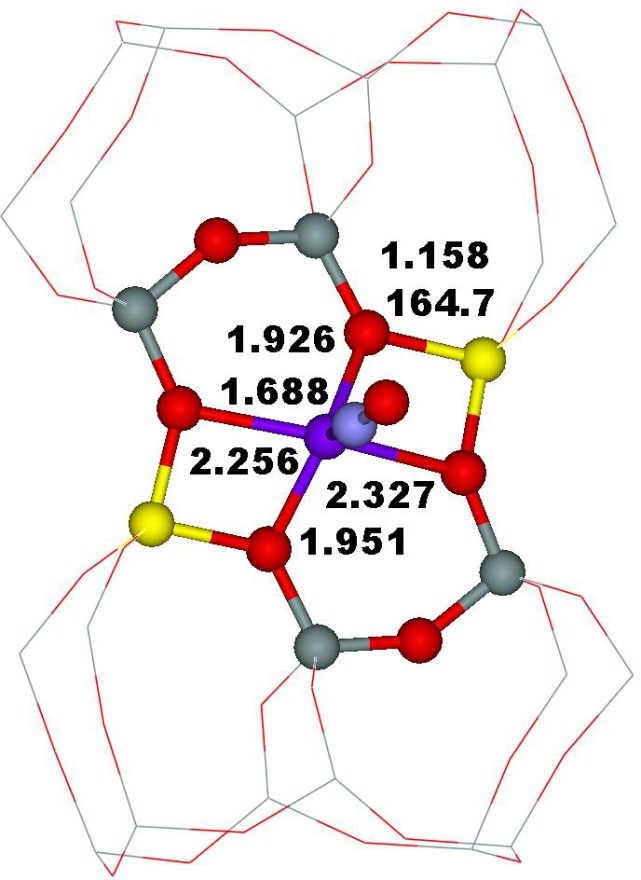
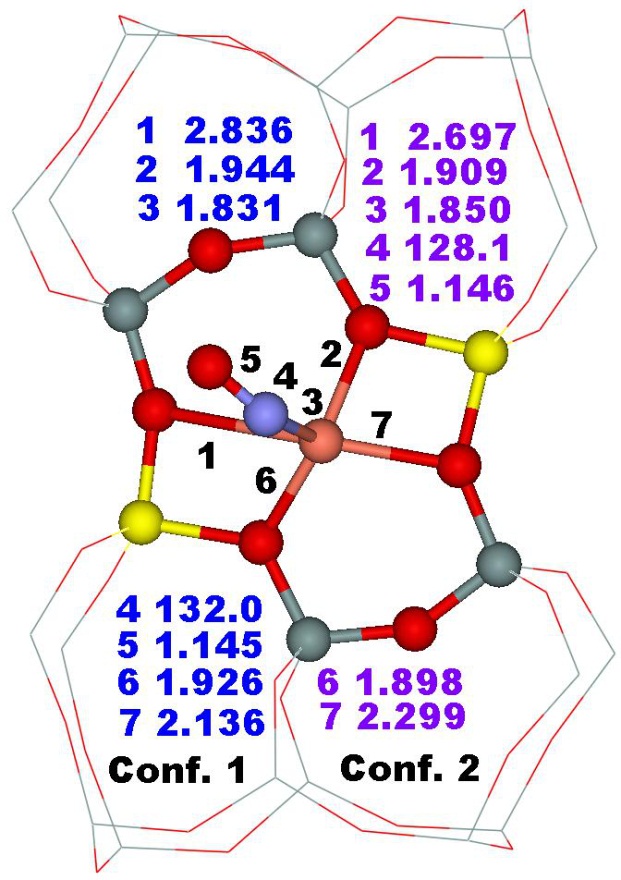
Figure 13. Optimized structures of the β–2 cationic site of NO–Co–ferrierite (left) and NO–Cu–ferrierite (right) after molecular dynamics simulations. The distances are in angstroms and the bond angle in degrees. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, nitrogen atom in blue, cobalt atom in violet, and copper atom in pink. Adapted Figure 8 from Ref.15
The presence of the band at 1864 cm−1 provides experimental evidence for the structural rearrangement of cationic sites since this IR band cannot be assigned to any site using the frequencies calculated for the structures which were not relaxed by employing molecular dynamics simulations. Our calculations show that the Al siting in the rings affects the local structure of the zeolite framework upon binding Cu(II) and Co(II). Moreover, we found that the binding of M(II) was the strongest in β–1, weaker in β–2, and the weakest in α. Therefore, the general tendency of Cu(II) and Co(II) accommodated in cationic sites to react is α > β–2 > β–1. These results clearly demonstrate the effects of the Al siting in the rings on the potential catalytic activity of the coordinated divalent cations and therefore justify the importance of the knowledge of the Al siting in the rings for understanding the catalytic properties of the divalent cation exchanged zeolites. Our results also reveal that FTIR spectroscopy of complexes of NO and divalent cation exchanged ferrierite can serve to identify the Al siting in the 6–rings forming the β–1 and β–2 cationic sites.
7.13. The organization of Al atoms in the framework Al–rich beta zeolites18
Two Al–rich and one Si–rich samples of the beta zeolite were studied by periodic DFT calculations including molecular dynamic simulations together with 27Al and 29Si (CP) MAS NMR, and FTIR of adsorbed acetonitrile–d3 and UV–vis spectroscopy of Co(II) cations as probes of close Al atoms. Our results show that in contrast to the Si–rich beta zeolites, the Al atoms in the Al–rich beta zeolites are mostly arranged in Al–O–Si–O−Al sequences with their Al atoms facing two different channels, which thus cannot bind bare divalent cations. Only Al atoms in Al–O–(Si–O)2–Al sequences in one ring and a minor fraction of Al–O–Si–O–Al sequences facing the same channel can balance bare divalent cations. The concentration of acid and redox sites in Al–rich beta zeolites, i.e., the potential catalytic active sites, is proportional to the increased Al content in the framework, but without marked change of their structure.
Our calculations of the (i) two types the β cationic sites with different location of the two Al atoms (i.e., Al(T6)–O–(Si–O)2–Al(T6) and Al(T4)–O–(Si−O)2–Al(T4)) and (ii) the α cationic site formed by Al(T3)–O–(Si–O)2–Al(T6) yielded the corresponding optimized structures (Figure 14).
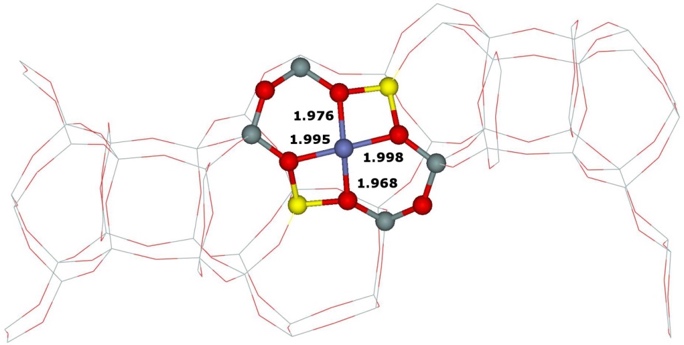
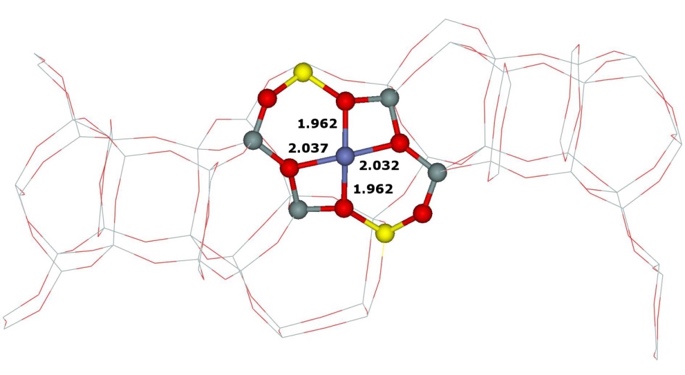
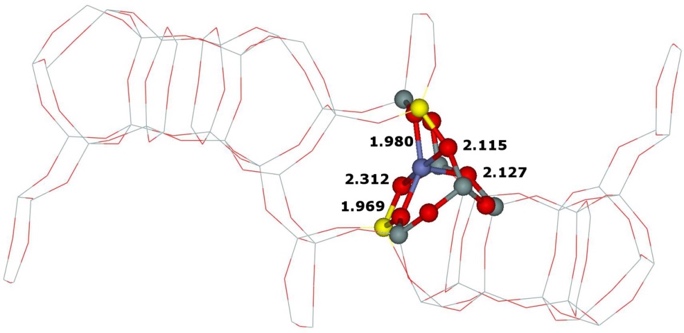
Figure 14. Optimized structures of the β cationic sites created by Al(T6)–O–(Si–O)2–Al(T6) (left), Al(T4)–O–(Si–O)2–Al(T4) (middle), and the α cationic site formed by Al(T3)–O–(Si–O)2–Al(T6) (right) after DFT molecular dynamics simulations. The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and cobalt atoms in blue. Adapted Figure 5 from Ref.18
The Co(II) cation accommodated in the β cationic site created by Al(T6)–O–(Si–O)2–Al(T6) is ligated to four oxygen atoms of two AlO4- tetrahedra, while the Co(II) cation in the β cationic site formed by Al(T4)–O–(Si–O)2–Al(T4) coordinates with two O atoms of two AlO4- tetrahedra and two O atoms of two SiO4 tetrahedra. Our prior computational studies of Fe(II)–FER,40 Co(II)–FER,15 and Cu(II)–FER15 showed significant rearrangements of the rings forming the α and β cationic sites upon binding the divalent cations. The reason why the β cationic site created by Al(T4)–O–(Si–O)2–Al(T4) site upon accommodation of Co(II) cations does not similarly rearrange in order for Co(II) to bind to four O atoms of two AlO4- tetrahedra, is most likely that the 6–ring forming this β cationic site creates a rigid double 6–ring (i.e., hexagonal prism) with another 6–ring. The stabilization energy obtained from the preferential binding of Co(II) to four O atoms of two AlO4- tetrahedra is most likely smaller than the energy needed to deform the structure of the double 6–ring to permit the preferential binding of Co(II).
7.14. Al organization and extra–framework sites of bare divalent cations in the TNU−9 zeolite20
The aluminum organization in the TUN framework of the TNU–9 zeolite was determined and the locations of the Al pairs forming the corresponding α and β cationic sites for bare divalent cations were suggested. Because the TNU–9 matrix is one of the most complex zeolites known, possessing 24 crystallographically distinguishable framework T sites and a highly complicated channel structure, the standard approach could not have been used. Therefore, we have developed a new significantly improved procedure that includes in addition to the standard methods also 27Al 3Q MAS NMR spectroscopy and extensive periodic DFT calculations, including molecular dynamics. This multi–spectroscopic and theoretical approach was shown to be a very powerful tool for analyzing the siting of aluminum pairs and divalent cations in the TNU–9 zeolite.
Our results reveal that 40 and 60% of aluminum atoms in the TUN framework are isolated single aluminum atoms and aluminum pairs (i.e., Al–O–(Si–O)2–Al sequences in one 6−ring forming cationic sites for divalent cations), respectively. Our study shows that Al–O–(Si–O)2–Al sequences are predominantly present in two types of 6–rings forming the corresponding α and β cationic sites for bare divalent cations.
We suggested based on our newly developed improved procedure without any prior knowledge of cationic sites for bare divalent cations from diffraction experiments the α and β cationic sites for bare divalent cations in the TNU–9 zeolite.
The α site represents the 6–ring formed from two 5–rings and is located on the TUN straight channel wall and connects two channel intersections. The two Al atoms are placed in positions diagonally across the 6−ring occupying either T4 and T24 or T10 and T22 (Figure 15).
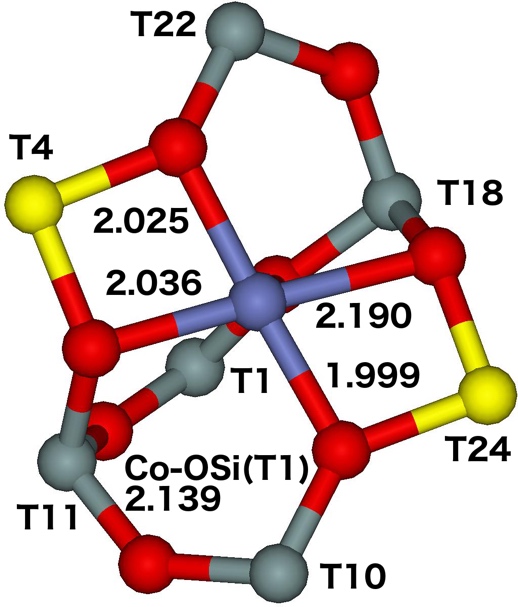
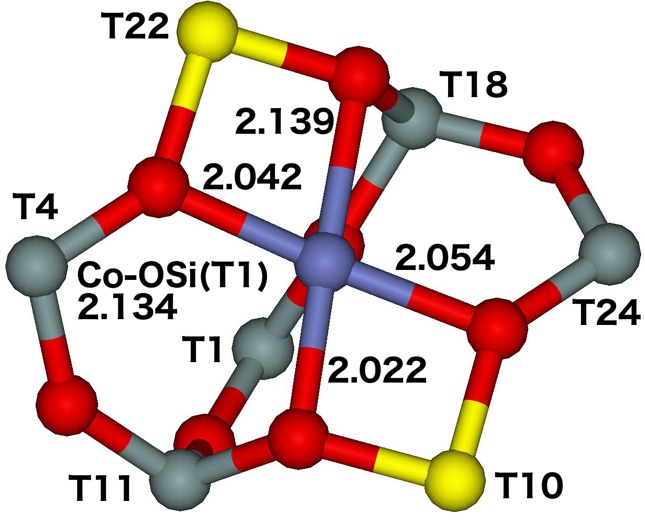
Figure 15. Optimized structures of the αT4T24 (left) and αT10T22 (right) cationic site after molecular dynamics simulations. The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and the cobalt atom in blue. Adapted Figure 13 from Ref.20
The Co(II) is placed in the plane of the 6–ring and is coordinated to four oxygens of two AlO4- tetrahedra and one oxygen of a SiO4 tetrahedron. The Co(II) cation exhibits an open coordination sphere and is easily accessible only from the straight channel; 9% of aluminum atoms (15% of Al pairs) are present in this site.
The β site is the main site for divalent cations and it accommodates at least 51% of Al atoms (85% of Al pairs). The near–planar βT11T4T9 site located at the channel intersection with the two aluminum atoms accommodated in T11 and T9 is the best candidate for the observed β cationic site. The near–planar βT8T10T11 site present also at the channel intersection with the two aluminum atoms placed in T11 and T8 is another less likely candidate. The optimized structures of both the sites are depicted in Figure 16.
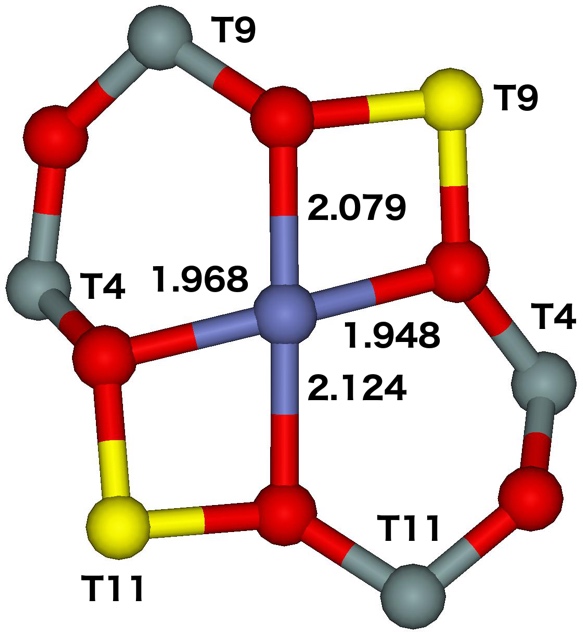
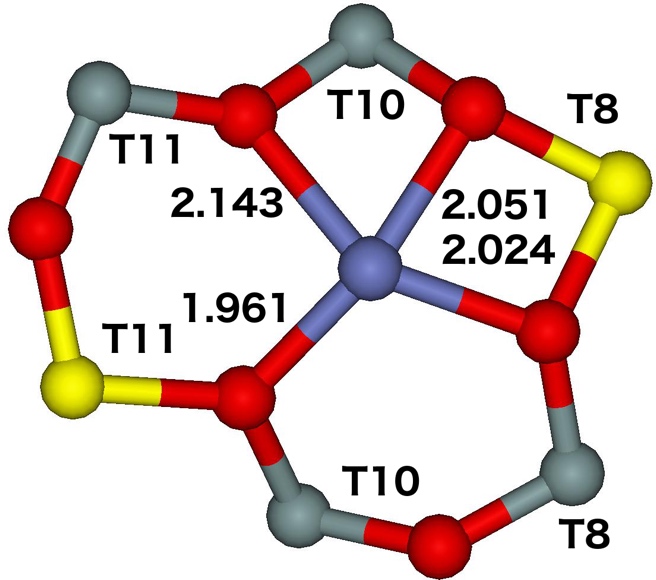
Figure 16. Optimized structures of the βT11T4T9 (left) and βT8T10T11 (right) cationic sites after molecular dynamics simulations. The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and the cobalt atom in blue. Adapted Figure 14 from Ref.20
The Co(II) is placed in the plane of this ring and is coordinated to four oxygen atoms of two AlO4- tetrahedra. The Co(II) accommodated in the β site are present at the channel intersections.
Based on the siting of Al–O–(Si–O)2–Al sequences forming the α and β cationic sites in the TNU–9 sample, our study has revealed that at least 60% of all the Al atoms (and most likely also the corresponding Brønsted acid sites) are located at the channel intersections. The location of Al pairs and thus of bare divalent cations in the TNU–9 zeolite is very similar to that in ZSM–5.
7.15. Al organization and extra–framework sites of bare divalent cations in the SSZ–13 zeolite3
SSZ–13 is a Si–rich (Si/Al > 5) small pore zeolite of the chabazite topology important for both acid and redox catalysis. The SSZ–13 matrix is not a pentasil–ring zeolite. We developed a new procedure involving 27Al (3Q) MAS NMR spectroscopy and extensive periodic DFT calculations with molecular dynamics, in addition to the standard methods developed for pentasil–ring zeolites based on bare Co(II) cations as probes monitored by FTIR spectroscopy and UV−vis spectroscopy. The location of the Al−O−(Si−O)2−Al and Al−O−(Si−O)3−Al sequences in the zeolite framework was determined (Al−O−Si−O−Al sequences are absent). 54% of the framework Al atoms correspond to Al−O−(Si−O)3−Al sequences which cannot form cationic sites for bare divalent cations but are able to accommodate divalent Co(II) hexaaqua complexes. Employing periodic DFT and 27Al (3Q) MAS NMR spectroscopy we determined that the corresponding Al−O−(Si−O)3−Al sequence is located in two double 6–ring cages with one Al located in the 4–ring connecting two double 6–ring units (Figure 17).
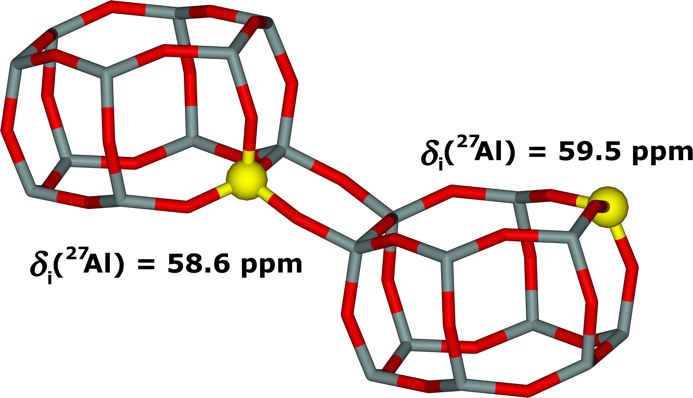
Figure 17. The structure corresponding to the most likely location of the close unpaired Al atoms (i.e., Al−O−(Si−O)3−Al) and their corresponding 27Al isotropic chemical shifts. Silicon atoms are in gray, oxygen atoms in red, and aluminum atoms in yellow. Adapted Figure 6F from Ref.3
Employing periodic DFT including extensive molecular
dynamics computations and 27Al (3Q) MAS NMR, UV–vis, and FTIR spectroscopies we determined that 35% of the framework
Al atoms could accommodate neither divalent Co(II) hexaaqua complexes nor bare
divalent cations. Furthermore, the siting of the Al atoms of the Al−O−(Si−O)2−Al
and Al−O−(Si−O)3−Al sequences forming four cationic sites for
divalent cations located in the 6–ring
(Al−O−(Si−O)2−Al), 8–ring
(Al−O−(Si−O)2−Al and Al−O−(Si− O)3−Al), and double 6–ring (Al−O−(Si−O)2−Al) was
determined (Figure 18).
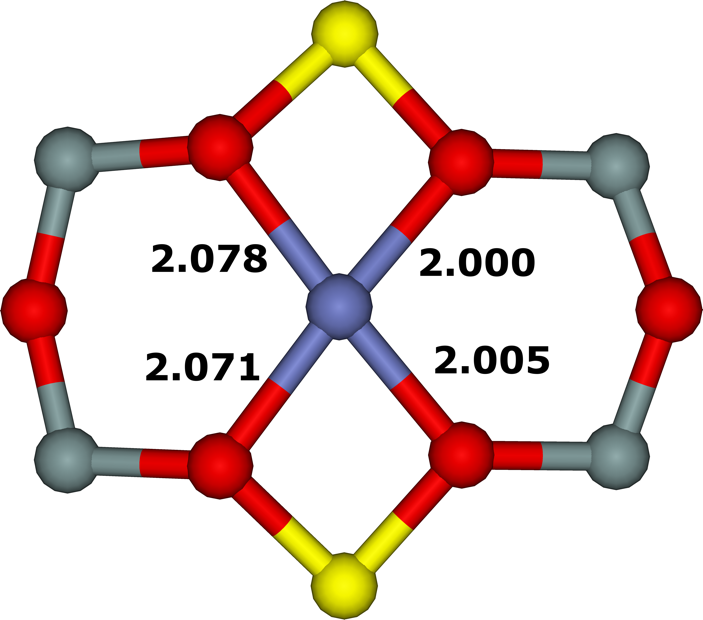
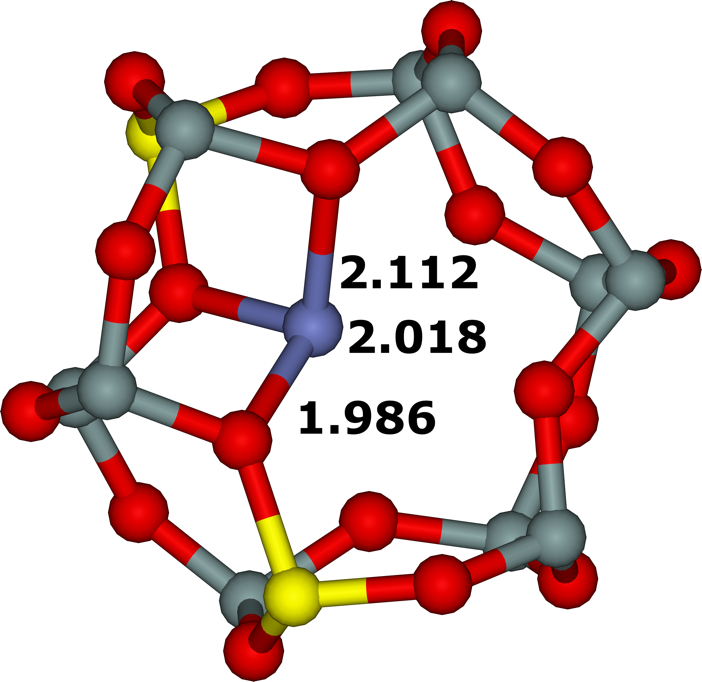
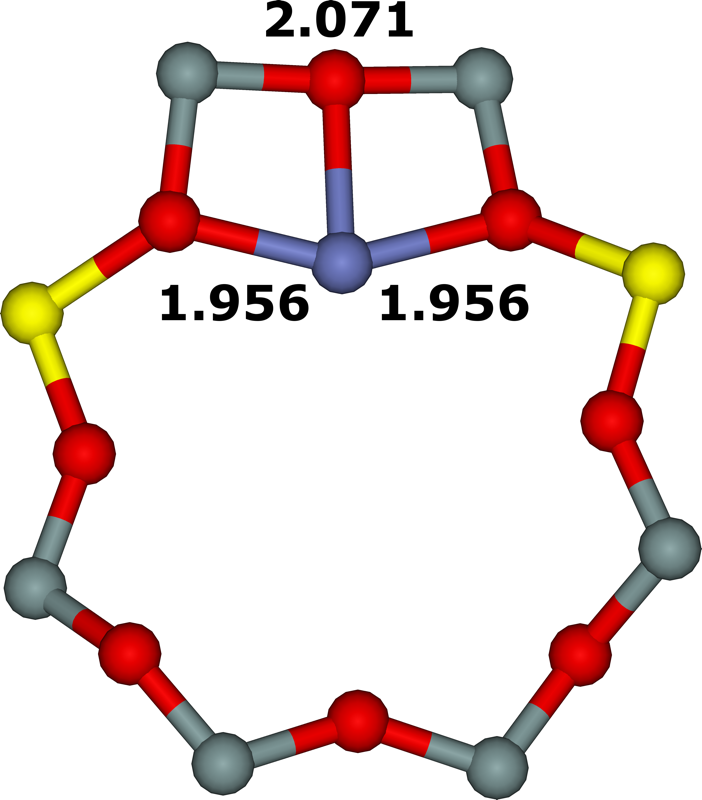
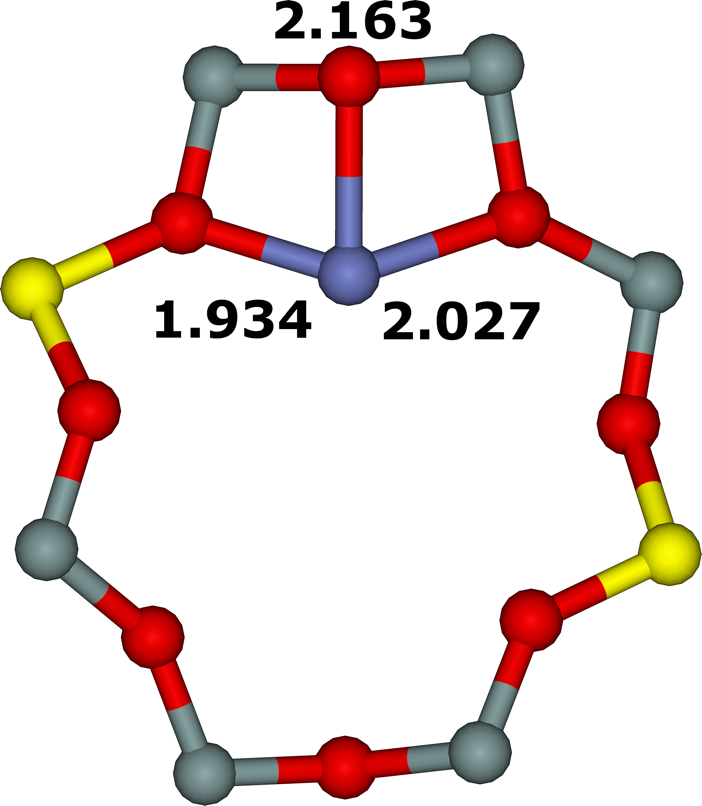
Figure 18. Optimized structures of the σ (extreme left), ω (left), τ (right), and τ3Si (extreme right) cationic sites after molecular dynamics simulations. The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and the cobalt atom in blue. Adapted Figure 3 from Ref.3
These Al pairs correspond to a minority of the Al framework atoms.
7.16. Modeling of Cu–oxo and Fe–oxo species in the beta zeolite42
The periodic DFT calculations including extensive molecular simulations were carried out to model structures whose general spectral features were observed during spectroscopic analysis of Fe and Cu exchanged beta zeolites (one Al–rich and one Si–rich), to obtain more complete information about their possible structures. Calculations were not performed to determine all of the possible configurations of the Cu and Fe species that could theoretically occur. The calculated dimeric species were coordinated to the framework rings with the known Al distribution in the Al–rich beta zeolite sample determined in our study,18 which indicated the predominant occurrence of close unpaired Al atoms (66%) linked with an Al−O−(Si−O)3−Al sequence.3 The Al−O−(Si−O)3−Al sequence located on a wall of the 12–ring channel was chosen to accommodate a dimeric site accessible for reactants. The calculated structures of possible dimeric metal–oxo species were modeled on the basis of previous studies of related Fe– and Cu–zeolite systems.
The activation of molecular oxygen over Cu zeolites was attributed in the literature to the dinuclear mono(μ–oxo)dicopper core ([Cu2(μ–O)]2+) and the bis(μ–oxo)dicopper core ([Cu2(μ–O)2]2+). Models of these two species were calculated. The results show that the distance between the two Cu atoms is 3.401 and 2.966 Å for the former and latter species, respectively (Figure 19).
Figure 19. Possible structures of Cu−oxo species located in accessible 12−ring channels and charge−compensated by a typical Al(T4)−O−(Si−O)3−Al(T5) sequence in the Al−rich beta zeolite. The optimized structures after the molecular dynamics simulations for (μ− oxo)dicopper ([Cu2(μ−O)]2+) (left) and bis(μ−oxo)dicopper ([Cu2(μ−O)2]2+) (right). The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and copper atoms in green. The spin state is denoted S. Adapted Figures 5a and 5b from Ref.42
The ground states were computed to be S = 2/2 and 4/2, respectively, and the Cu oxidation states were 2 and 3, respectively. There is no “α−oxygen−like” atom166 in the structure. The bridging oxygen atoms could be responsible for the oxidative properties of the system. All our attempts to optimize the structure of the (μ−peroxo)dicopper species analogous to complex 6 in Ref.167 failed. The optimizations yielded structures identical to bis(μ−oxo)dicopper ([Cu2(μ−O)2]2+). Therefore, we assumed that the (μ−peroxo)−dicopper structure does not correspond to a minimum on the potential energy surface. This result is consistent with the fact that complex 6 in ref.167 is not thermally stable.
As the partial oxidation of methane over Fe−exchanged zeolites was associated with various diiron core structures, five models for the dinuclear iron cations with μ−oxo and μ−peroxo ligands were calculated. The first Fe model (Figure 20, left) yielded a structure in which the Fe1 atom features two oxygen atoms forming double bonds to Fe1 (the calculated bond lengths are 1.603 and 1.609 Å).
Figure 20. Possible structures of Fe−oxo species located in accessible 12−ring channels. The optimized structures after the DFT molecular dynamics simulations for the first Fe model charge−compensated by an Al(T3)−(Si−O)2−Al(T3) sequence and Al(T2) (left) and the second Fe model balanced by an Al(T3)−(Si−O)2−Al(T3) sequence and Al(T4) (right). The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and iron atoms in blue. The spin state is denoted S. Adapted Figures 6a and 6b from Ref.42
These two O atoms resemble the α oxygen atom166 and it is probable that they could be responsible for the oxidative properties of the studied system. The distance between the two Fe atoms is 3.494 Å and therefore they are connected by a μ−oxo bridge. The ground state is calculated to correspond to S = 7/2. The formal oxidation states of Fe1 and Fe2 are 6 and 3, respectively. Therefore, the Fe1 atom corresponds to the Fe atom of ferrate(VI) (Fe(VI)O42−).168 The computational results regarding the second Fe model (Figure 20, right) indicate a structure in which the Fe1 atom possesses one oxygen atom forming double bonds to Fe1 (the calculated bond length is 1.634 Å). In addition, this O atom resembles the α oxygen atom166 and could be linked to the oxidative properties of the system. The Fe1 and Fe2 atoms are connected by a μ−peroxo bridge due to a larger distance between the two Fe atoms (4.481 Å). The ground state is computed to be related to S = 9/2. The formal oxidation states of Fe1 and Fe2 are 4 and 3, respectively.
By employing DFT calculations, Li et al. found that two close [FeO]+ species readily form one bis(μ−oxo)diiron structure in ZSM−5.140 This structure was calculated as the third Fe model (Figure 21, top, left).
Figure 21. Possible structures of Fe−oxo species located in accessible 12−ring channels. The optimized structures after the DFT molecular dynamics simulations the third Fe model (left), the fourth Fe model (middle), and the fifth Fe model (right) with all of the three models charge−compensated by Al(T4)−(Si−O)3−Al(T5). The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and iron atoms in blue. The spin state is denoted S. Adapted Figures 6c, 6d, and 6e from Ref.42
The two Fe atoms are calculated to be 2.634 Å from each other. The formal oxidation state of the two Fe atoms is 3. The spin state S = 8/2 was calculated to be the ground state and is more stable by 7.0 kcal·mol−1 than the state with S = 10/2. Therefore, the bis(μ−oxo)diiron structure in the studied beta zeolite was calculated to be an intermediate−spin species. There is no “α oxygen−like” atom166 in the structure and therefore the bridging O atoms could be responsible for the oxidative properties of the system.
The (μ−oxo)(μ−peroxo)diiron structure was calculated as the fourth model (Figure 21, top, right). It is the precursor of the (μ−oxo)diiron which is calculated as the fifth model (Figure 21, bottom, left). The (μ−oxo)diiron species features two oxygen atoms forming double bonds to the two Fe atoms (the calculated bond lengths are 1.633 and 1.630 Å). These two O atoms resemble the α−oxygen atom166 and they could be responsible for the oxidative properties of the system. The distances between the two Fe atoms were calculated to be equal to 3.234 and 3.432 Å for the fourth and fifth Fe models, respectively. The calculations indicated that the ground state is connected with S = 8/2 for both the models. The formal oxidation state of the two Fe atoms is 4. The fifth Fe model is calculated to be more stable than the fourth model and the third Fe model by 4 and 13 kcal/mol, respectively.
Our computational results indicate that these copper−oxo and iron−oxo species could exist in the investigated beta zeolite and lead to indications of their reactivity.
7.17. Structure of the distant binuclear Fe(II),40, 43-44 Co(II),41, 44 Mn(II)44 cationic sites in ferrierite
Accommodation of Fe(II), Co(II), and Mn(II) cations in two adjacent β−2 cationic sites of the ferrierite zeolite were investigated using periodic DFT calculations including molecular dynamics simulations. The two adjacent β−2 cationic sites with M(II) form the distant binuclear cationic sites. These computations led to the same rearrangements of the cationic sites upon binding of bare divalent cations to oxygen atoms of the rings forming the cationic sites as in the case of isolated β−2 cationic sites in ferrierite accommodating Fe(II),40 Co(II),15 and Cu(II)15 cations. The optimized structures (Figure 22) served for calculations of the formation of the α–oxygen atoms using either N2O40-41 or O243-44 molecules.
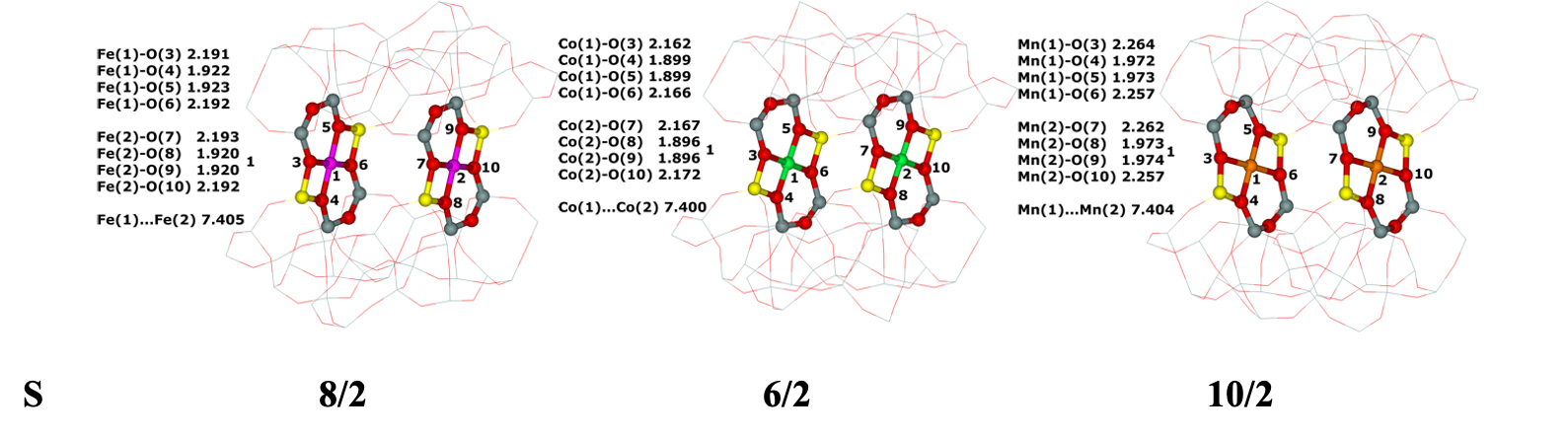
Figure 22. Optimized structures after the molecular dynamics simulations for the two adjacent β−2 cationic sites in ferrierite accommodating Fe(II) (left), Co(II) (middle), and Mn(II) (right). The spin state is denoted S. Adapted Figures 3A, 1A, and 2A from Ref.44
7.18. Structure of the distant binuclear Fe(II) cationic sites in the beta zeolite45
We investigated employing periodic DFT calculations including molecular dynamics simulations the accommodation of Fe(II) cations in two opposite β cationic sites of the beta zeolite. The two opposite β cationic sites are across the 12–ring channel. Two different Al sitings in the 6–rings forming these opposite β cationic sites were studied: (i) Al(T5), Al(T5), Al(T6), and Al(T6) (the BEAβT5βT6 model) and (ii) Al(T7), Al(T7), Al(T8), and Al(T8) (the BEAβT7βT8 model). The two opposite β cationic sites with M(II) are a candidate structure for the formation of the distant binuclear cationic sites. The optimized structures of the BEAβT5βT6 and BEAβT7βT8 models after the MD calculations are shown in Figure 23.
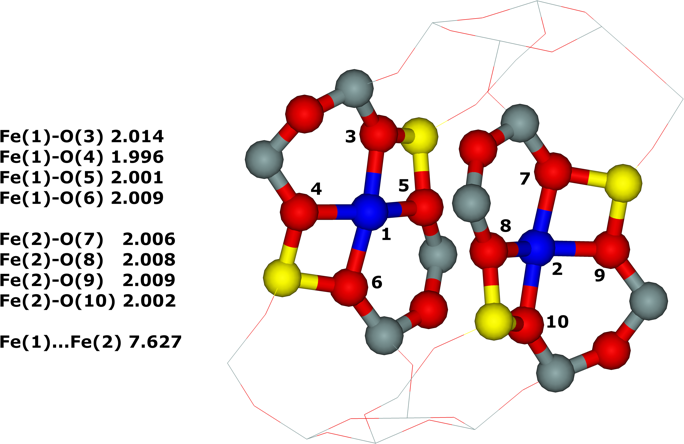
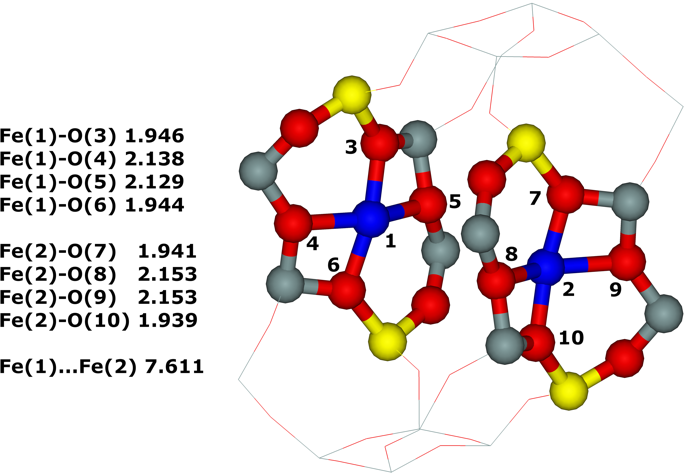
Figure 23. Optimized structures of the beta zeolite after the molecular dynamics simulations for two opposite β cationic sites across the 12–ring channel with two different Al sitings in the 6–rings forming these opposite β cationic sites: the BEAβT5βT6 model (left) and the BEAβT7βT8 model (right). The distances are in Å. Silicon atoms are in gray, aluminum atoms are in yellow, iron atoms in are blue, and oxygen atoms are in red. Adapted Figures 3 (right) and 4 (right) from Ref.45
Both the models of the beta zeolite moderately rearrange (ΔE = 1−4 kcal/mol), and only the BEAβT5βT6 model possesses the proper coordination of Fe(II) to four oxygen atoms of two AlO4- tetrahedra before and after the MD calculations. Conversely, Fe(II) in BEAβT7βT8 ligates to two O atoms of two AlO4- tetrahedra and two O atoms of two SiO4 tetrahedra before and after the MD calculations. The same results were obtained for Co(II)–exchanged beta zeolites in our prior study.18 The reason why the BEAβT7βT8 model does not rearrange in order for Fe(II) to bind to four O atoms of two AlO4- tetrahedra is most likely that the 6–rings forming these β cationic sites create a rigid double 6–ring (i.e., hexagonal prism) with another 6–ring. The stabilization energy obtained from the proper binding of Fe(II) to four O atoms of two AlO4- tetrahedra is most likely smaller than the energy needed to deform the structure of the double 6–ring to permit the proper binding of Fe(II).
The β cationic sites of the beta zeolite are located on the wall of the 12–ring channel. Two opposite β cationic sites are present at the opposite sides of the wall of the 12–ring channel. The mutual geometrical position of the two cationic sites and the Fe···Fe distance are shown in Figure 23 for both the BEAβT5βT6 and BEAβT7βT8 models.
7.19. Structure of the distant binuclear Fe(II) cationic sites in mordenite45
The binding of Fe(II) cations in two adjacent β cationic sites of mordenite was studied using periodic DFT calculations including molecular dynamics simulations. Two models with two Fe(II) cations accommodated in Al(T1)−Si(T1)−Si(T3)−Si(T3)−Al(T1) and Al(T3)−Si(T3)−Si−(T1)−Si(T1)−Al(T3) sequences in the 8−rings forming the β cationic sites were employed in the MORβT1βT1 and MORβT3βT3, respectively, models. The two remaining MOR models based on the other two possible Al sitings in the 8–ring were not studied and can be ruled out as candidate structures for the formation of the distant binuclear cationic sites since our preliminary calculations revealed that either the Fe cations were coordinated in a different ring or the coordination of the Fe(II) cations is pseudo–tetrahedral with a closed coordination sphere and therefore not meeting experimentally observed spectra of Co(II) cations in the β cationic site of mordenite.9
The optimized structures of the MORβT1βT1 and MORβT3βT3 models before and after the MD calculations are shown in Figures 24 and 25, respectively.
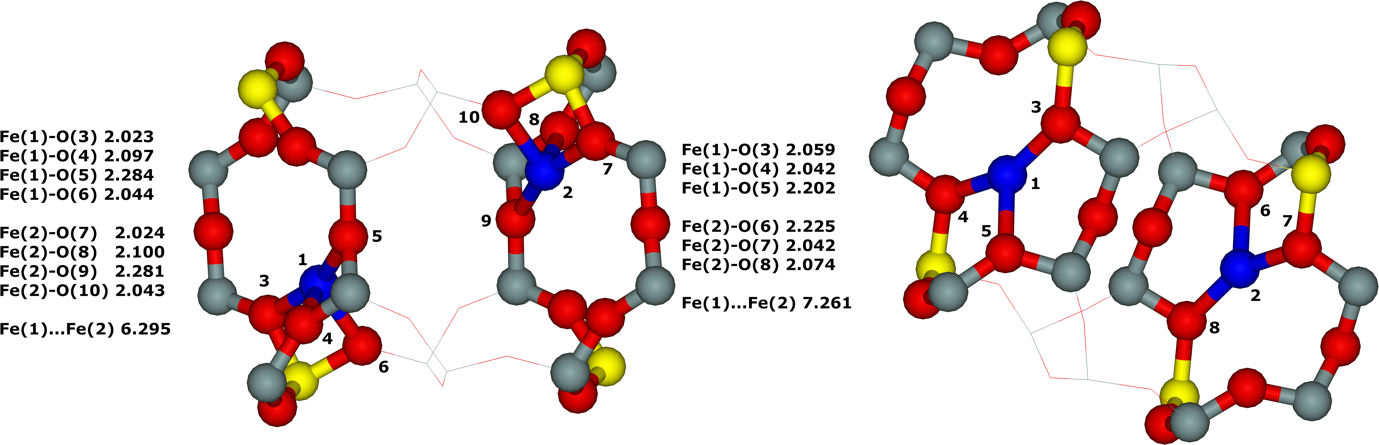
Figure 24. Optimized structures of mordenite before (left) and after (right) the DFT molecular dynamics simulations for two adjacent βT1 (the MORβT1βT1 model) sites. The distances are in Å. Silicon atoms are in gray, aluminum atoms are in yellow, iron atoms are in blue, and oxygen atoms are in red. Adapted Figure 6 from Ref.45
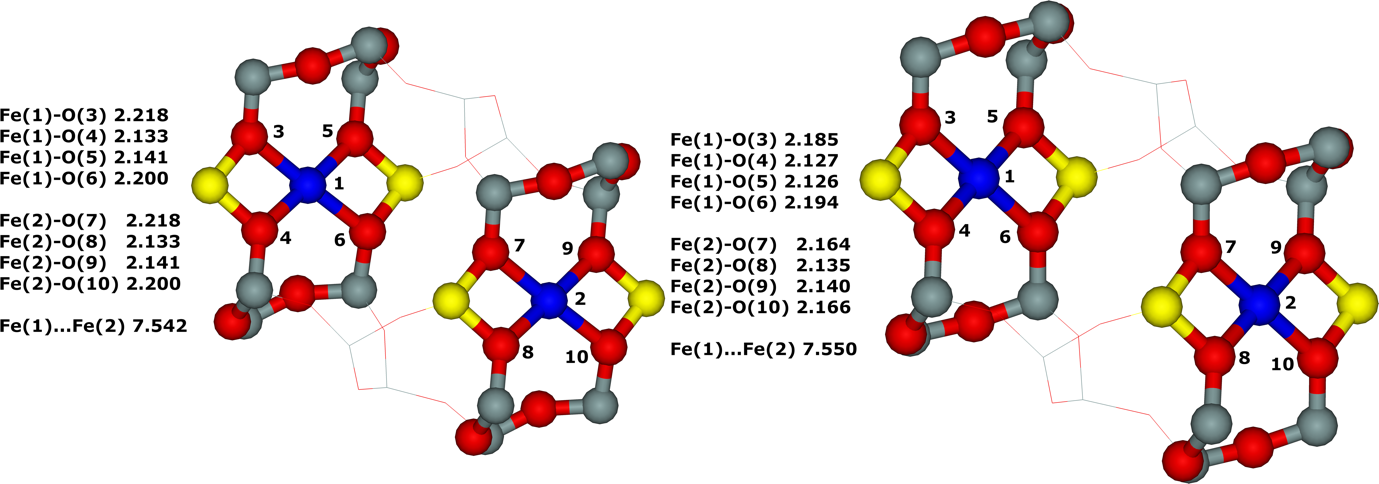
Figure 25. Optimized structures of mordenite before (left) and after (right) the DFT molecular dynamics simulations for two adjacent βT3 (the MORβT3βT3 model) sites. The distances are in Å. Silicon atoms are in gray, aluminum atoms are in yellow, iron atoms are in blue, and oxygen atoms are in red. Adapted Figure 7 from Ref.45
The results of our study show that the MORβT1βT1 model greatly rearranges (ΔE = 11 kcal/mol), while the MORβT3βT3 model only moderately changes (ΔE = 3 kcal/mol) by the molecular dynamics calculations. The Fe(II) cations in the former coordinate to two oxygen atoms of an AlO4- tetrahedron and two oxygen atoms of a SiO4 tetrahedron before the MD calculations, while they ligate to two oxygen atoms of two AlO4- tetrahedra and one oxygen atom of a SiO4 tetrahedron after the MD calculations. Conversely, the latter features the proper coordination of Fe(II) to four oxygen atoms of two AlO4- tetrahedra before and after the MD calculations.
The β cationic sites in the mordenite structure are located at the bottom of the mordenite pocket. They are accessible only through 8–rings from the main channel. The mutual geometrical position of the two cationic sites and the Fe···Fe distance are shown in Figures 24 and 25 for both the MORβT1βT1 and MORβT3βT3 models.
8. N2O decomposition40, 46, 128
We used periodic DFT calculations including molecular dynamics together with multiple spectroscopies40 to study the N2O decomposition46 over Fe–ferrierite, Fe–ZSM–5, and Fe–beta zeolite. The results40 reveal that the distant binuclear Fe(II) sites in Fe–ferrierite are responsible for the superior activity of this catalyst in the N2O decomposition in the low temperature region. Two Fe(II) cations coordinated in two adjacent β cationic sites of Fe–ferrierite form the active sites for the N2O decomposition.
The calculated Fe–Fe distance of the active site is 7.4 Å. The formation of the active sites results from a combination of (1) a suitable topology of the ferrierite framework and (2) an appropriate distribution of Al in the distinguishable T sites of the ferrierite framework as well as concentration of Al in these T sites. Both 6–rings forming the two adjacent β cationic sites must contain two Al atoms each (four Al atoms in total). Figure 26 shows the initial N2O complex in which N2O is adsorbed by the N terminal atom to one of the two Fe(II) cations accommodated in the two adjacent β–2 cationic sites forming the distant binuclear Fe(II) sites which are the active sites for the N2O decomposition.
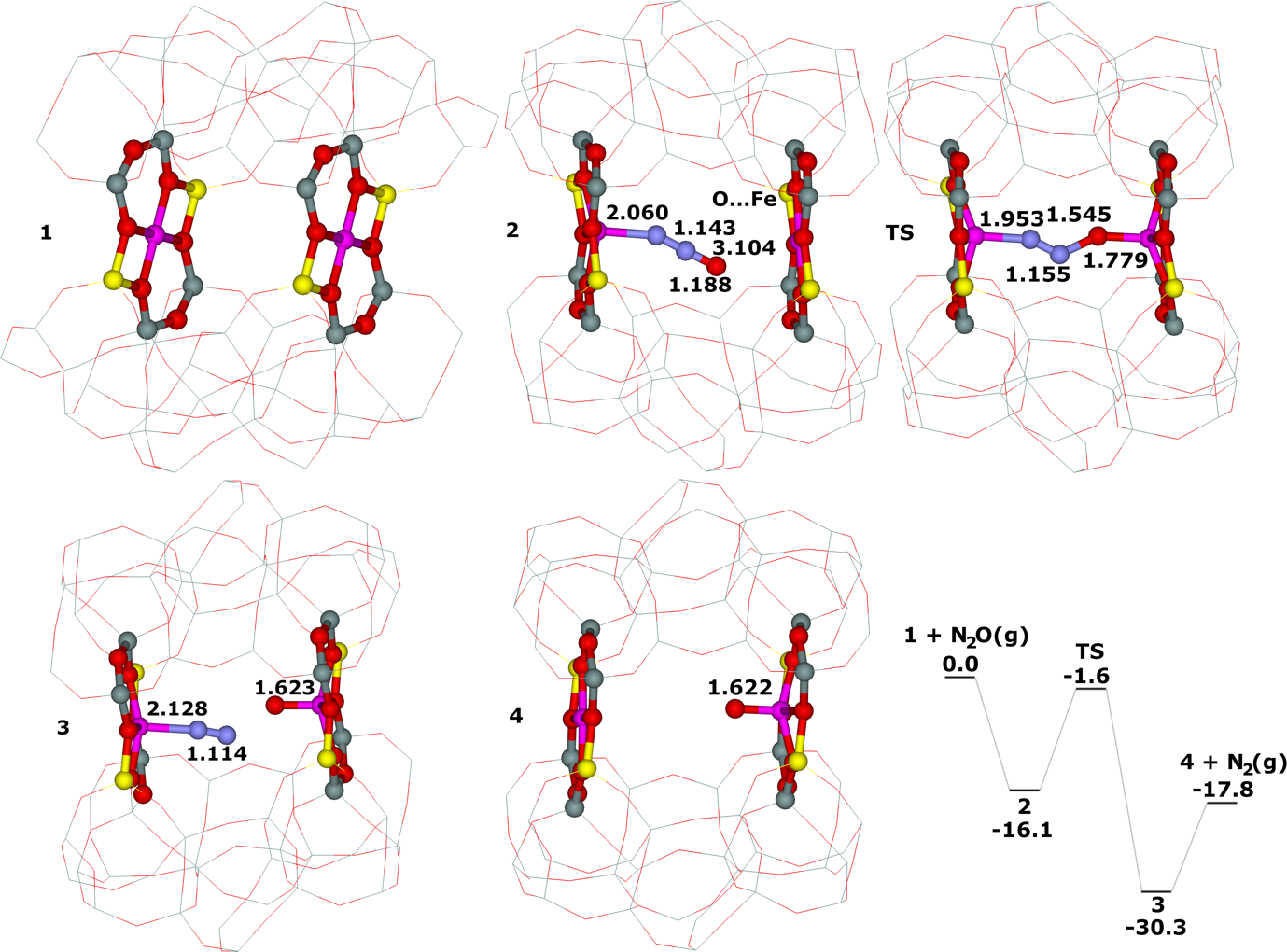
Figure 26. Optimized structures of the β sites of Fe−ferrierite (1) after molecular dynamics simulations (top, left), the [Fe…NNO Fe] complex (2) formed in the two adjacent β sites (top, middle), the [Fe−N−N−O−Fe] transition state (TS) created in the two adjacent β sites (top, right), the [Fe…NN O═Fe] intermediate (3) formed in the two adjacent β sites (bottom, left), the [Fe O═Fe] product (4) created in the two adjacent β sites (bottom, middle). The distances are in angstroms. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, iron atom in violet, and nitrogen atoms in blue. Schematic energy profile (in kcal mol-1) of the formation of the α−oxygen (bottom, right). The data taken from Ref.41
Figure 26 shows that N2O adsorbs by the N terminal atom on one Fe(II) cation. The O atom of the adsorbed N2O is well positioned to oxidize the other Fe(II) cation to yield the α−oxygen atom166 and adsorbed N2 after the N−O bond cleavage. The calculated barrier is 14.5 kcal/mol. This value indicates that the oxidation of Fe(II) to give the α−oxygen should be very facile.
Two opposite β cationic sites of the Si–rich Fe–beta zeolite can also form an active site with two collaborating Fe(II) cations. However, there are significant differences with respect to ferrierite. The distribution and concentration of Al in the rings with the geometry of the β cationic site lead to a very low probability of occurrence of the active sites with two cooperating Fe(II) cations. This is the reason for the superior activity of Fe–ferrierite relative to Fe–beta.
Two close α cationic sites of the Fe(II) exchanged ZSM–5 zeolite coordinating two collaborating Fe(II) cations could compose the active site. However, the geometrical arrangement of two adjacent α cationic sites is very distant from that of two adjacent β sites in ferrierite. Also, the probability of a formation of these active sites is low due to an unfavorable Al distribution and Al concentration in the rings forming the α cationic sites.
The results of our experimental and theoretical investigation reveal that the order of the activity of the Fe(II) exchanged zeolites is as follows: Fe–ferrierite >> Fe–beta > Fe–ZSM–5.
Our combined experimental and theoretical investigation using an isotope exchange with N218O revealed that zeolite–framework O atoms were involved in the formation of O2 molecules during the N2O decomposition catalyzed by Fe–ferrierite. We suggested plausible mechanisms of the isotope exchange.128
9. Formation of the α−oxygen atom on the distant binuclear Co(II) sites in Co–ferrierite from N2O41
Our study reveals for the first time that the α−oxygen with notable oxidation properties can be prepared not only on a Fe(II)−zeolite but also on a zeolite exchanged with two other divalent cations: Co(II) and Ni(II). The variability of the type of the cation forming the distant binuclear active sites can potentially allow the tuning of the corresponding catalytic properties. The α−oxygen is formed over all the three M(II)−ferrierite samples (M = Co(II), Ni(II), and Fe(II)) by the abstraction of the oxygen atom from N2O by the distant binuclear M(II) sites. The M(II) cations are accommodated in two adjacent β−2 cationic sites of M(II)−ferrierite and create the distant binuclear active sites responsible for the formation of the α−oxygen. The α−oxygen is then able to selectively oxidize methane mainly to methanol at room temperature. Only the distant binuclear Co(II)...Co(II) structures and not isolated Co(II) cations are active in the formation of the α−oxygen while both the types of Fe(II) cations are potent to yield the α−oxygen.
The formation for the α−oxygen was calculated for Co(II) (Figure 27) and Fe(II), but not for Ni(II) for technical reasons.
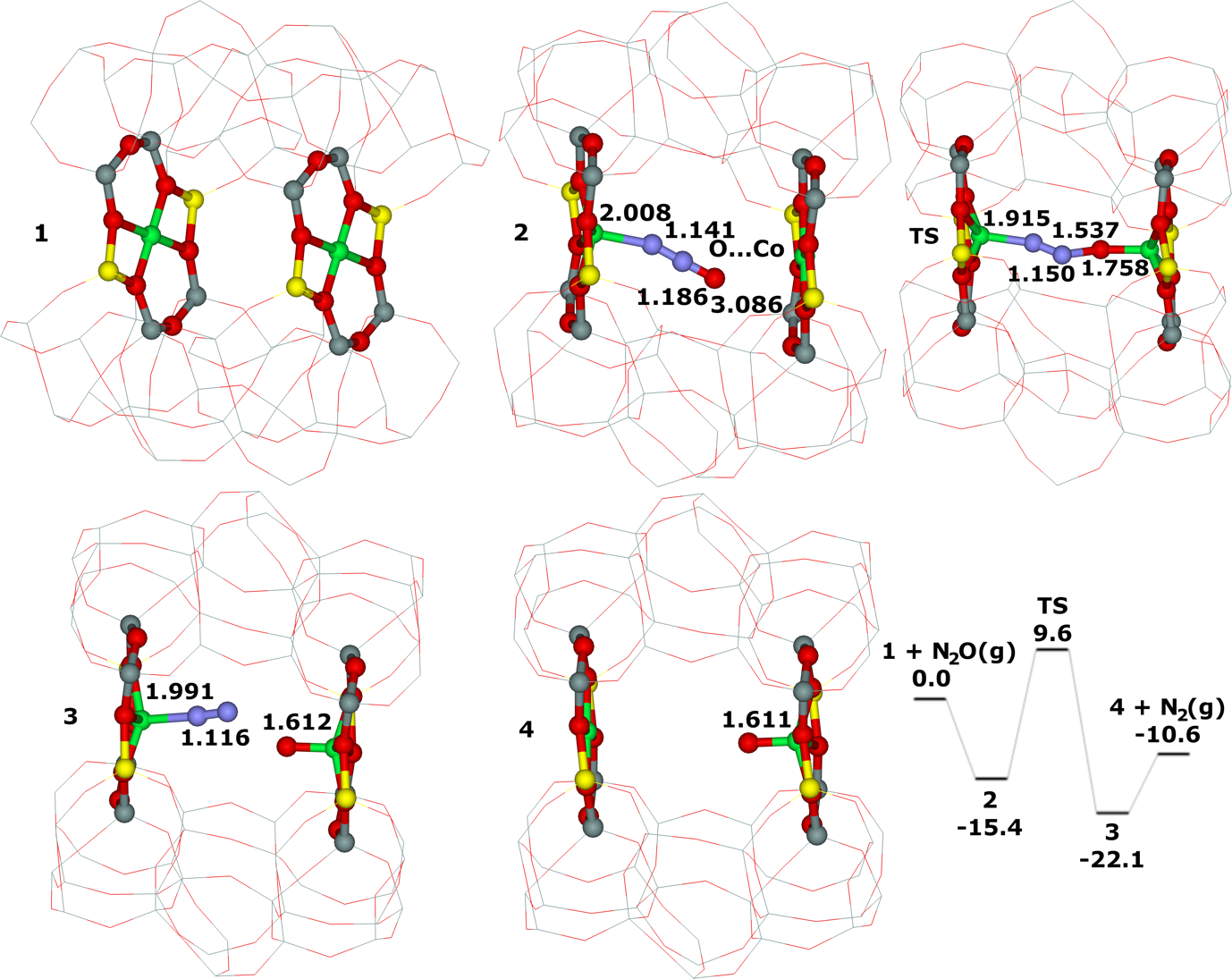
Figure 27. Optimized structures of the β sites of Co−ferrierite (1) after molecular dynamics simulations (top, left), the [Co…NNO Co] complex (2) formed in the two adjacent β sites (top, middle), the [Co−N−N−O−Co] transition state (TS) created in the two adjacent β sites (top, right), the [Co…NN O═Co] intermediate (3) formed in the two adjacent β sites (bottom, left), the [Co O═Co] product (4) created in the two adjacent β sites (bottom, middle). The distances are in angstroms. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, cobalt atom in green, and nitrogen atoms in blue. Schematic energy profile (in kcal mol-1) of the formation of the α−oxygen (bottom, right). Adapted Figure 1 from Ref.41
Figure 27 shows that N2O adsorbs by the N terminal atom on one Co(II) cation. The O atom of the adsorbed N2O is well positioned to oxidize the other Co(II) cation to yield the α−oxygen and adsorbed N2 after the N−O bond cleavage. The calculated barrier is 25.0 kcal/mol. This value indicates that the oxidation of Co(II) to give the α−oxygen should be facile but significantly more sluggish than the same reaction step on Fe(II)−ferrierite (i.e., the barrier of 14.5 kcal/mol).
10. Splitting dioxygen at room temperature to form the active α−oxygen for methane oxidation at room temperature43
Activation of dioxygen is a basic enzymatic reaction of living organism while mimicking this process over artificial inorganic systems represents a great challenge. The activation of dioxygen gained significance as a possible key for the usage of methane. Methane as the main component of natural gas became abundant because of the development of the shale gas technology. Nevertheless, until now, productions of energy (electricity and heat) and hydrogen represent the main utilization of the methane production. Therefore, the transformation of methane to liquid products representing energy carriers and chemical production platforms is in high demand. The selective oxidation of methane to methanol is suggested to be an encouraging way of a methane−to−liquid transformation. However, only the selective oxidation of methane by dioxygen is economically feasible and represents a promising system of the utilization of methane with an enormous industrial impact.
10.1. The role of distant binuclear M(II) sites in M−ferrierite40-41, 43-44, 53
Our studies showed that M(II) (M = Co(II), Ni(II), and Fe(II)) cations exchanged in the ferrierite zeolite form distant binuclear cationic structures which significantly facilitate the abstraction of the oxygen atom from N2O to yield the highly active α−oxygen on the M(II) cation. The M(II) cations forming these species are located in two adjacent extra−framework cationic β sites. The calculated distance of the two M(II) cations is ca. 7.4 Å. In contrast to isolated M(II) cations, the binuclear M(II) species can arrange a four−electron reaction. These distant binuclear M(II) sites in M(II)−ferrierite exhibit a resemblance in geometry and the oxidation state with iron active sites of methane monooxygenases; however, the distance between the two Fe cations in the enzymes is less than half of that in M(II)−ferrierite. This raised a question if distant binuclear M(II) structures were able to cleave dioxygen and form a pair of the distant α−oxygen atoms on the two M cations which afterward can oxidize methane to methanol. Employing periodic DFT calculations in tandem with Mössbauer (only for Fe) and FTIR spectroscopies and stoichiometric reaction tests we answered this question, see Sections 10.2. and 10.3.
10.2. Splitting dioxygen over Fe(II)−ferrierite43
Our periodic DFT calculations of Fe(II)−ferrierite reveal that two Fe(II) cations forming distant binuclear cationic structures can indeed cleave dioxygen to form two α−oxygen atoms. Figure 28 shows the calculated mechanism of the cleavage.
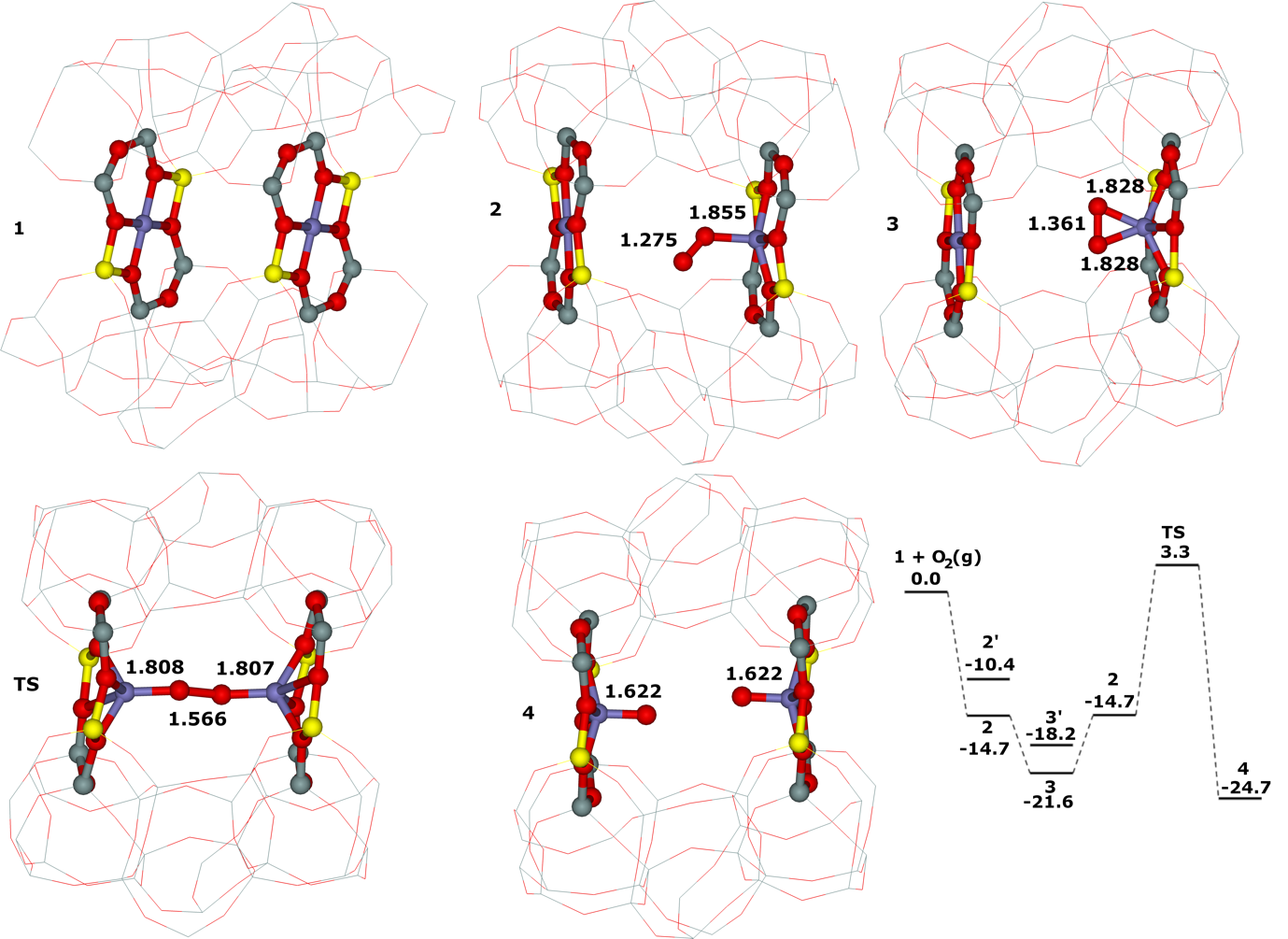
Figure 28. Optimized structures of the two adjacent β sites of Fe−ferrierite (1) after molecular dynamics simulations (top, left), the monodentate [Fe OOmono…Fe] complex (2) formed in the two adjacent β sites (top, middle), the bidentate [Fe OObi…Fe] complex (3) formed in the two adjacent β sites (top, right), the [Fe−O−O−Fe] transition state (TS) created in the two adjacent β sites (bottom, left), the [Fe═O O═Fe] product (4) created in the two adjacent β sites (bottom, middle). The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and iron atoms in blue. Schematic energy profile (in kcal mol-1) of the formation of the [Fe═O O═Fe] product (bottom, right). Adapted Figure 1 from Ref.43
An O2(g) molecule that is in a triplet state adsorbs on one of the Fe(II) cations to yield a [Fe OOmono...Fe]′ monodentate complex 2′ with the O2 moiety in a triplet state. 2′ either undergoes a spin crossover to give a [Fe OOmono...Fe] monodentate complex 2, which has the O2 moiety in a singlet state, or rearranges its structure to form a [Fe OObi...Fe]′ bidentate complex 3′. In the latter case, a spin change occurs and a [Fe OObi...Fe] bidentate complex 3 with the O2 moiety in a singlet state is yielded. The oxidation occurs from the most stable bidentate complex 3, which rearranges to the less stable monodentate complex 2. The adsorbed O2 moiety of 2 is better positioned for the interaction with the other Fe(II) located in the adjacent β site. Subsequently, dioxygen is cleaved via a [Fe−O−O−Fe] transition state TS to yield a [Fe═O O═Fe] complex 4 in a concerted manner. Both Fe in 4 are oxidized to form a pair of the distant α−oxygen atoms. The reaction energy of the reaction from 1 + O2(g) to give 4 is −24.7 kcal/mol. The calculated barrier of the cleavage of dioxygen is 24.9 kcal/mol, indicating that the oxidation should be facile but substantially more sluggish than the oxidation of the same Fe(II)−ferrierite by N2O (i.e., the barrier of 14.5 kcal/mol41).
Our Mössbauer and FTIR experiments and stoichiometric reaction tests show that methane can be oxidized by dioxygen over distant binuclear Fe(II) species stabilized in an aluminosilicate matrix. Both the formation of the α−oxygen atoms and the oxidation of methane to methanol occur at room temperature. This outcome indicates a breakthrough in the development of the technology of the transformation of methane to liquid products representing energy carriers and chemical production platforms. Nevertheless, the application in the chemical industry requires a further development of a long−term stable system with a high activity in the methane conversion.
10.3. Splitting dioxygen over M(II)−ferrierite (M = Co(II) and Mn(II))44, 53
Our periodic DFT computations for Co(II)−ferrierite and Mn(II)−ferrierite reveal that our breakthrough in the activation of dioxygen is not limited exclusively to Fe(II) cations but the ability of dioxygen splitting represents a general property of the distant binuclear M(II) centers capable of the M(II) to M(IV) redox cycle with the M...M distance of ca 7.4 Å stabilized in M–ferrierite. Our results reveal that the distant binuclear M(II) (M(II) = Co(II) and Mn(II)) sites located in two adjacent β sites of ferrierite can split dioxygen and form a pair of the α–oxygen species which were reported to be very active in the oxidation of methane to methanol. Figures 29 and 30 show the calculated mechanism of the cleavage for Co(II)−ferrierite and Mn(II)−ferrierite, respectively.
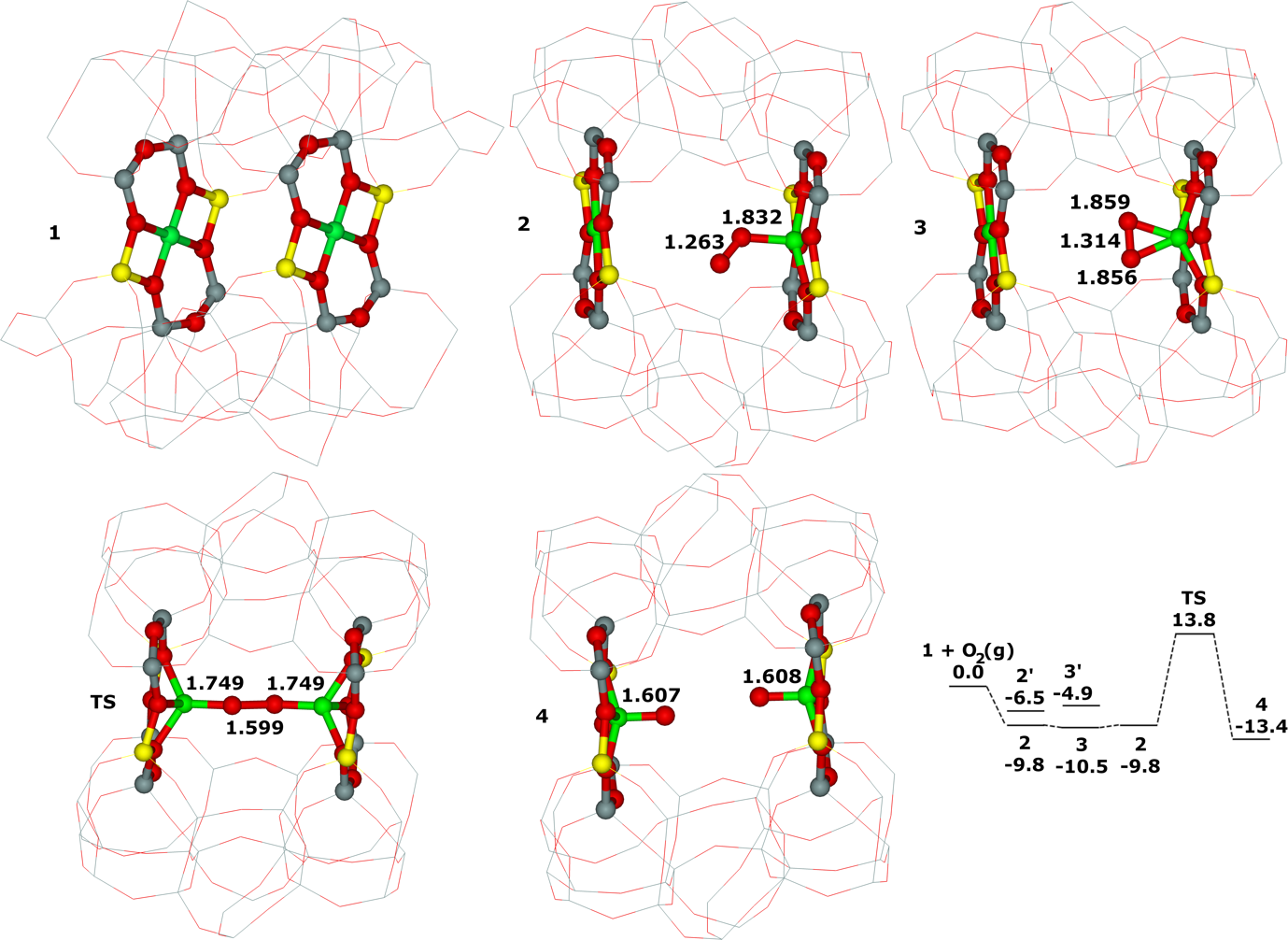
Figure 29. Optimized structures of the two adjacent β sites of Co−ferrierite (1) after molecular dynamics simulations (top, left), the monodentate [Co OOmono…Co] complex (2) formed in the two adjacent β sites (top, middle), the bidentate [Co OObi…Co] complex (3) formed in the two adjacent β sites (top, right), the [Co−O−O−Co] transition state (TS) created in the two adjacent β sites (bottom, left), the [Co═O O═Co] product (4) created in the two adjacent β sites (bottom, middle). The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and cobalt atoms in green. Schematic energy profile (in kcal mol-1) for the spin state S = 6/2 (2′ and 3′ are S = 8/2) of the formation of the [Co═O O═Co] product (bottom, right). Adapted Figure 1 from Ref.44
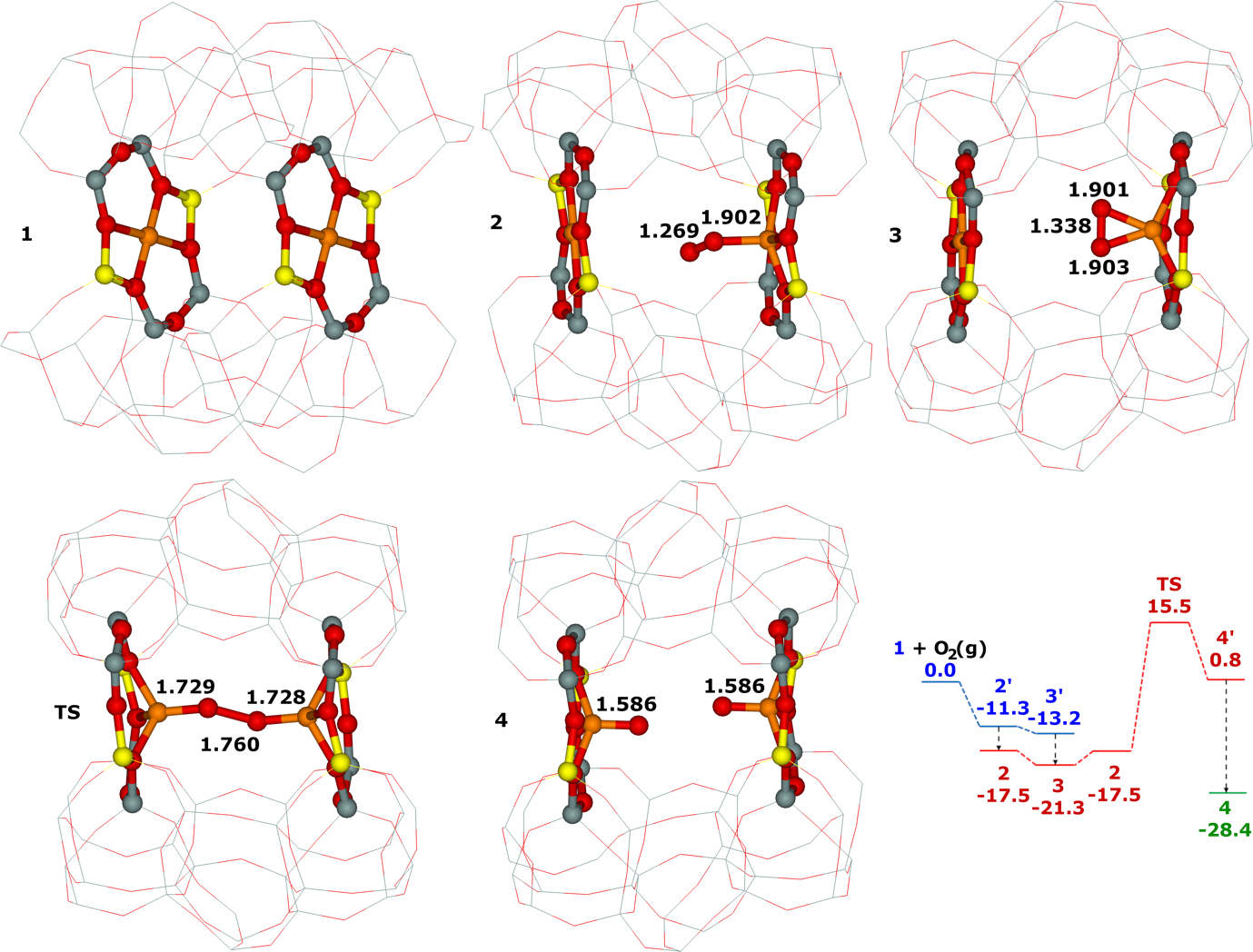
Figure 30. Optimized structures of the two adjacent β sites of Mn−ferrierite (1) after molecular dynamics simulations (top, left), the monodentate [Mn OOmono…Mn] complex (2) formed in the two adjacent β sites (top, middle), the bidentate [Mn OObi…Mn] complex (3) formed in the two adjacent β sites (top, right), the [Mn−O−O−Mn] transition state (TS) created in the two adjacent β sites (bottom, left), the [Mn═O O═Mn] product (4) created in the two adjacent β sites (bottom, middle). The distances are in Å. Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and manganese atoms in orange. Schematic energy profile (in kcal mol-1) for the spin state S = 10/2 blue profile, S = 8/2 red profile, and S = 6/2 green profile of the formation of the [Mn═O O═Mn] product (4) (bottom, right). Adapted Figure 2 from Ref.44
The calculated mechanisms of splitting dioxygen for Co(II)–ferrierite and Mn(II)–ferrierite are generally the same as that for Fe(II)–ferrierite. The calculated barrier of 24.3 kcal/mol for Co(II)–ferrierite is essentially the same as that for Fe(II)–ferrierite (24.9 kcal/mol) while the barrier for Mn(II)–ferrierite is significantly higher (36.8 kcal/mol) indicating that the cleavage of dioxygen over Mn(II)–ferrierite is expected to be more sluggish.
Our FTIR experiments and stoichiometric reaction tests confirmed the creation of a pair of the α–oxygen species for Co(II)–ferrierite, Mn(II)–ferrierite, and Ni(II)–ferrierite at room temperature and the subsequent oxidation of methane to methanol at room temperature as well. The formation for the α−oxygen was not calculated for Ni(II) for technical reasons.
10.4. Splitting dioxygen over Fe(II)−zeolites other than ferrierite45
The effects of the local arrangement of distant binuclear Fe(II) centers and framework topology on the ability to split dioxygen to form a pair of the α–oxygen atoms was investigated in a subsequent study. The goal of the study was to answer a question whether the low barriers of the cleavage of dioxygen result from the unique topology of the ferrierite zeolite and the Al organization (especially the Al siting in the rings forming the cationic sites) in the ferrierite used or if the activity regarding splitting dioxygen represents a general property of the distant binuclear Fe(II) centers stabilized in the aluminosilicate matrix. If the latter is true, it can represent a highly promising base for the development of exceptionally active systems with higher concentrations of the active sites for the direct oxidation of methane. Moreover, splitting dioxygen over the distant binuclear Fe(II) centers located at the opposite sides of the wall of larger channels potentially opens the possibility of using the α–oxygen atoms also for the direct oxidation of bulkier molecules with a restricted access to the α–oxygen atoms through 8–rings (i.e., via the ferrierite side channel). It should be noted that syntheses of zeolites of different topologies with the controlled Al organization would require a long–term highly complex and demanding research. This clearly evidences that a computational approach represents the only way permitting to answer the questions raised above. We employed periodic DFT calculations to investigate the effect of (i) the Al siting in the rings forming the cationic sites, (ii) the distance, and (iii) the mutual geometrical position of the rings accommodating Fe(II) on the activity of the distant binuclear Fe(II) sites in splitting dioxygen. The ferrierite, mordenite, beta, and A zeolites were employed for this purpose.
Figure 31 shows the calculated mechanism of the cleavage of dioxygen over Fe(II)−beta.
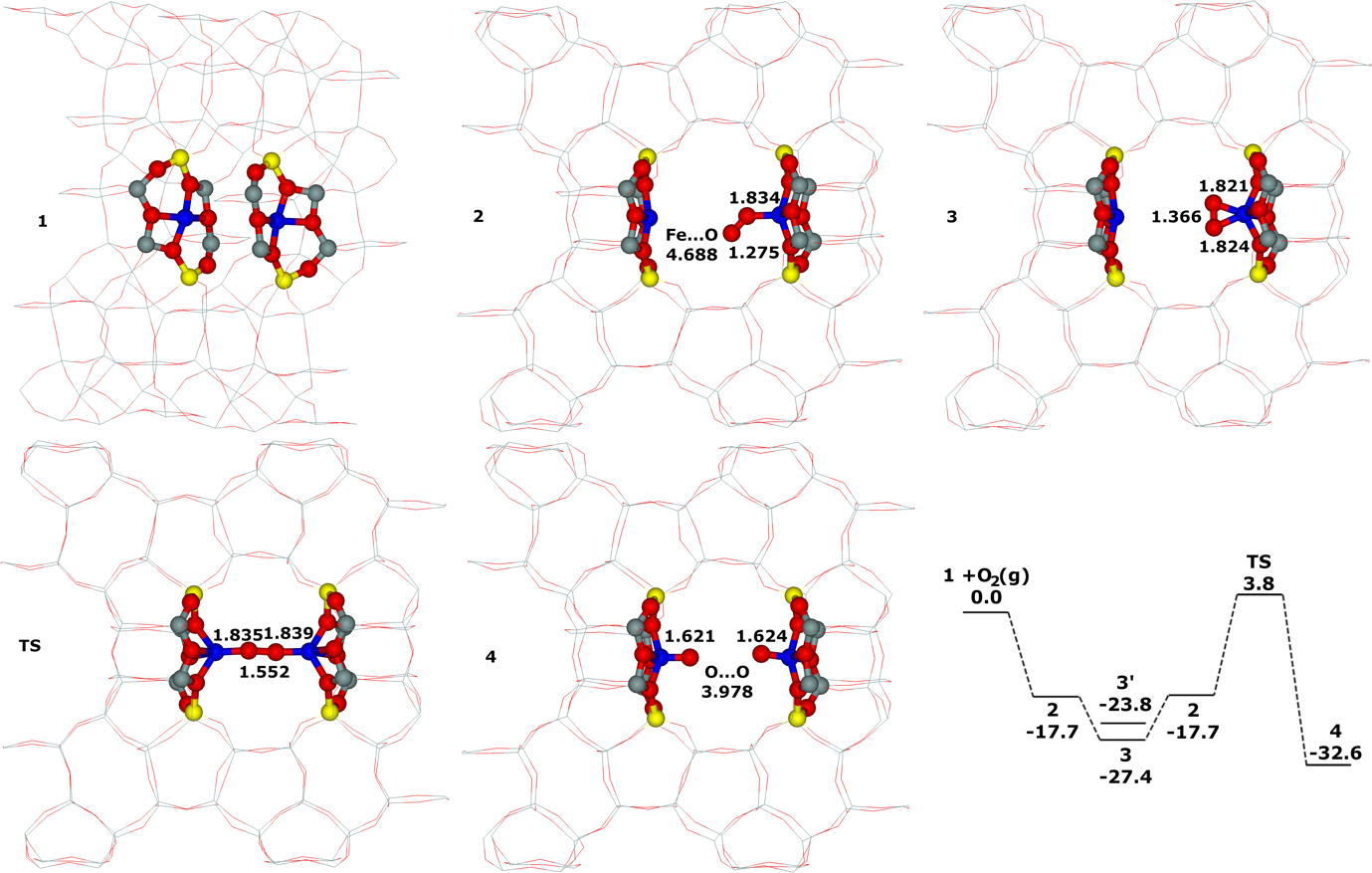
Figure 31. Optimized structures for the two opposite β(Al(T7) and Al(T7)) and β(Al(T8), and Al(T8)) sites (i.e. the BEAβT7βT8 model) of the Fe−beta zeolite (1) after molecular dynamics (MD) simulations (top, left), the monodentate [Fe OOmono···Fe] complex (2) (top, middle), the bidentate [Fe OObi···Fe] complex (3) (top, right), the [Fe−O−O−Fe] transition state (TS) (bottom, left), and the [Fe═O O═Fe] product (4) (bottom, middle). The distances are in Å. Silicon atoms are in gray, oxygen atoms are in red, aluminum atoms are in yellow, and iron atoms are in blue. Schematic energy profile (in kcal mol-1) for the formation of the [Fe═O O═Fe] product (4) (bottom, right). Adapted Figure 10 from Ref.45
Our results reveal that the distant binuclear Fe(II) sites with suitable parameters accommodated in various zeolites can split dioxygen and form a pair of the α–oxygen atoms able to oxidize methane to methanol. Therefore, the ability to cleave dioxygen represents a general property of the distant binuclear Fe(II) centers stabilized in aluminosilicate matrices, thus suggesting the possibility of developing Fe–zeolite–based systems for the dioxygen activation for direct oxidations using various zeolite matrices. The suitable parameters are found to be the two cationic sites forming the distant binuclear Fe(II) centers have to (i) face each other, (ii) be parallel, and (iii) be axial. (iv) The Fe···Fe distance has to lie in a narrow range from ca. 7 to ca. 8 Å (ca. 7−ca. 10 Å for the distance between the two rings (forming the corresponding cationic sites) in empty zeolites since this distance is equal or larger than the Fe···Fe distances).
10.5. Splitting dioxygen over M(II)−zeolites
Based on our studies of M(II)–ferrierite43-44, 53 (M(II) = Fe(II), Co(II), Mn(II) and Ni(II)) and Fe(II)–zeolite45 (zeolite = ferrierite, mordenite, beta, and A zeolites) we can generalize that the distant binuclear M(II) centers capable of the M(II) to M(IV) redox cycle and with the suitable parameters (Section 10.4) can cleave dioxygen to form a pair of the α–oxygen atoms able to oxidize methane to methanol. This suggests the possibility of developing M(II)–zeolite based tunable systems for the activation of dioxygen for direct oxidations using various transition metal cations in various zeolite matrices.
10.6. Reactivity of the distant binuclear Fe(II) centers54
We used periodic DFT calculations to investigate the detailed mechanism of the reduction of Fe(IV)═O of Fe–ferrierite by dihydrogen. The findings attained for the Fe(IV)═O centers of pairs of the distant α–oxygen atoms were compared with those obtained for the isolated Fe(IV)═O sites. The oxidation of dihydrogen, which is the simplest oxidation reaction, was chosen for comparison of the activity of both the types of Fe(IV)═O. Moreover, the reduction of Fe(IV)═O by dihydrogen represents the basis of the H2−TPR (temperature–programmed reduction by dihydrogen) method, which is one of the essential methods used for the characterization of cationic species in zeolites.
The results of our study reveal that (i) there is no direct simultaneous reduction of both the Fe(IV)═O centers of pairs of the distant α–oxygen atoms, (ii) one Fe(IV)═O site of a pair of the distant α–oxygen atoms is reduced by a molecule of dihydrogen to yield adsorbed water on the Fe(II) cation while the other Fe(IV)═O site is intact, and subsequently, one of the hydrogen atoms of the adsorbed water is abstracted by the other intact α–oxygen atom to yield two Fe(III)−OH groups, which are afterward reduced by another molecule of dihydrogen to give two water molecules each adsorbed on one Fe(II) cation, (iii) an isolated Fe(IV)═O site is reduced by the same mechanism as one Fe(IV)═O site of a pair of the distant α–oxygen atoms, and (iv) lower reducibility of the Fe(IV)═O centers of pairs of the distant α–oxygen atoms relative to the isolated Fe(IV)═O sites. The obtained results clearly evidence (and most likely can be generalized for other molecules than dihydrogen as well) that the proximity of the other Fe(IV)═O site in the confined reaction space of the zeolite cavity can dramatically change the behavior of both the cooperating α–oxygen atoms and the reaction mechanism over Fe(IV)═O sites of a pair of the distant α–oxygen atoms can differ from that over isolated Fe(IV)═O sites.
11. Conclusions
Our work significantly contributed to the determination of the organization of Al atoms in the framework of industrially important Si–rich zeolite catalysts. The Al organization is a key property and its knowledge is required to evaluate the potential of a zeolite for individual catalytic reactions.
Our development of the bare zeolite framework model permitted the employment of realistic models for our DFT calculations of the local geometry of AlO4- tetrahedra in fully hydrated, cation–containing silicon–rich zeolites, and subsequently the evaluation of 27Al isotropic chemical shifts which allowed for the first time the partial determination of the Al siting for the ZSM–5 zeolite and the full resolution of the Al siting for the ferrierite zeolite. We showed that the Al siting is neither random nor controlled by a simple rule but it depends on the conditions of the zeolite synthesis. Our achievements clearly demonstrate the power of the high–resolution 27Al MAS NMR experiment combined with DFT calculations and also support the bare zeolite framework model adopted in our calculations.
We developed a method which uses periodic DFT calculations and 7Li and 23Na MAS NMR experiments for the analysis of the siting of monovalent Li+ and Na+, respectively, cations in extra–framework cationic sites in Si–rich ferrierite zeolites. These monovalent cations in dehydrated zeolite frameworks can be used as additional probes of the Al organization. Li+ cations as probes can determine the Al siting while Na+ cations as probes can clearly identify the ring accommodating the Na+ cation, while the Al siting in that ring can be determined only for special cases. Our studies of the siting of monovalent Li+ and Na+ are the first ones which determine the local structure of Li+ and Na+, respectively, extra–framework cationic sites in a Si–rich zeolite with no knowledge of the Li+ and Na+, respectively, cationic sites from diffraction experiments.
Employing 27Al solid–state NMR and FTIR experiments in tandem with DFT calculations we determined for the first time that framework Al Lewis sites correspond to Al atoms tricoordinated to the zeolite framework, and furthermore, we suggested a plausible mechanism of their formation in the beta zeolite.
Using only the power of DFT calculations we determined the local structure of cationic sites for bare divalent transition metal cations (i.e., Fe(II), Co(II), Mn(II), and Cu(II)) and showed that these cations upon binding to cationic sites induced a rearrangement of the local structure of the zeolite framework. The local structure of cationic sites for bare divalent transition metal cations cannot be determined by diffraction methods.
The knowledge of (i) the organization of Al atoms in the framework of Si–rich zeolites and (ii) the local structure of cationic sites for bare divalent transition metal cations in these zeolites was a necessary condition of our (i) identification of the distant binuclear cationic sites and (ii) theoretical prediction of their ability to split dioxygen afterward confirmed by experiments.
We recognized the distant binuclear cationic sites using only theoretical modeling of the N2O decomposition over Fe–zeolites. The distant binuclear Fe(II) sites are responsible for the superior activity of Fe–ferrierite in the N2O decomposition in the low temperature region. Afterward, we predicted using solely the power of periodic DFT calculations that these distant binuclear Fe(II) sites were able to split dioxygen to yield pairs of the distant α–oxygen atoms able to oxidize methane to methanol at room temperature. This theoretical prediction was subsequently confirmed by experiments and thus splitting dioxygen was discovered. This achievement represents a breakthrough in oxidation catalysis.
Theoretical modeling further clearly showed that this
breakthrough in the activation of dioxygen was not limited exclusively to
Fe(II) cations and the structure of the ferrierite zeolite. The ability of
dioxygen splitting represents a general property of the distant binuclear M(II)
centers accommodated in a zeolite matrix if the M(II) cations (i) are capable
of the M(II) to M(IV) redox cycle and (ii) have suitable structural parameters.
Our achievements open the possibility of developing highly active and selective
systems employed for the direct oxidation of (i) methane to methanol and (ii)
other organic compounds to valuable oxidation products.
References
(1) Catlow, C. R.; Davidson, M.; Hardacre, C.; Hutchings, G. J. Philos. Trans. R. Soc. A 2016, 374, 20150089.
(2) Sklenak, S.; Dedecek, J.; Li, C. B.; Wichterlova, B.; Gabova, V.; Sierka, M.; Sauer, J. Angew. Chem., Int. Ed. 2007, 46, 7286-7289.
(3) Mlekodaj, K.; Dedecek, J.; Pashkova, V.; Tabor, E.; Klein, P.; Urbanova, M.; Karcz, R.; Sazama, P.; Whittleton, S. R.; Thomas, H. M.; Fishchuk, A. V.; Sklenak, S. J. Phys. Chem. C 2019, 123, 7968-7987.
(4) Dedecek, J.; Sobalik, Z.; Wichterlova, B. Catal. Rev. Sci. Eng. 2012, 54, 135-223.
(5) Dedecek, J.; Tabor, E.; Sklenak, S. ChemSusChem 2019, 12, 556-576.
(6) Knott, B. C.; Nimlos, C. T.; Robichaud, D. J.; Nimlos, M. R.; Kim, S.; Gounder, R. ACS Catal. 2018, 8, 770-784.
(7) Baerlocher, C.; McCusker, L. B. Database of zeolite structures: http://www.iza-structure.org/databases/ (accessed April 25, 2022).
(8) Pashkova, V.; Sklenak, S.; Klein, P.; Urbanova, M.; Dedecek, J. Chem. Eur. J. 2016, 22, 3937-3941.
(9) Dedecek, J.; Wichterlova, B. J. Phys. Chem. B 1999, 103, 1462-1476.
(10) Kaucky, D.; Dedecek, J. I.; Wichterlova, B. Microporous Mesoporous Mater. 1999, 31, 75-87.
(11) Dedecek, J.; Kaucky, D.; Wichterlova, B. Microporous Mesoporous Mater. 2000, 35-6, 483-494.
(12) Dedecek, J.; Kaucky, D.; Wichterlova, B. Chem. Commun. 2001, 970-971.
(13) Dedecek, J.; Kaucky, D.; Wichterlova, B.; Gonsiorova, O. Phys. Chem. Chem. Phys. 2002, 4, 5406-5413.
(14) Gabova, V.; Dedecek, J.; Cejka, J. Chem. Commun. 2003, 1196-1197.
(15) Sklenak, S.; Andrikopoulos, P. C.; Whittleton, S. R.; Jirglova, H.; Sazama, P.; Benco, L.; Bucko, T.; Hafner, J.; Sobalik, Z. J. Phys. Chem. C 2013, 117, 3958-3968.
(16) Sobalik, Z.; Sazama, P.; Dedecek, J.; Wichterlova, B. Appl. Catal., A 2014, 474, 178-185.
(17) Klein, P.; Dedecek, J.; Thomas, H. M.; Whittleton, S. R.; Pashkova, V.; Brus, J.; Kobera, L.; Sklenak, S. Chem. Commun. 2015, 51, 8962-8965.
(18) Sazama, P.; Tabor, E.; Klein, P.; Wichterlova, B.; Sklenak, S.; Mokrzycki, L.; Pashkkova, V.; Ogura, M.; Dedecek, J. J. Catal. 2016, 333, 102-114.
(19) Klein, P.; Pashkova, V.; Thomas, H. M.; Whittleton, S. R.; Brus, J.; Kobera, L.; Dedecek, J.; Sklenak, S. J. Phys. Chem. C 2016, 120, 14216-14225.
(20) Karcz, R.; Dedecek, J.; Supronowicz, B.; Thomas, H. M.; Klein, P.; Tabor, E.; Sazama, P.; Pashkova, V.; Sklenak, S. Chem. Eur. J. 2017, 23, 8857-8870.
(21) Klein, P.; Dedecek, J.; Thomas, H. M.; Whittleton, S. R.; Klimes, J.; Brus, J.; Kobera, L.; Bryce, D. L.; Sklenak, S. J. Phys. Chem. C 2022, 126, 10686-10702.
(22) Dedecek, J.; Wichterlova, B. Phys. Chem. Chem. Phys. 1999, 1, 629-637.
(23) Dedecek, J.; Capek, L.; Wichterlova, B. Appl. Catal., A 2006, 307, 156-164.
(24) Zhang, L. W.; Zhang, H. K.; Chen, Z. Q.; Ning, Q.; Liu, S. Y.; Ren, J.; Wen, X. D.; Li, Y. W. Catal. Sci. Technol. 2019, 9, 7034-7044.
(25) Wang, S.; He, Y.; Jiao, W. Y.; Wang, J. G.; Fan, W. B. Curr. Opin. Chem. Eng. 2019, 23, 146-154.
(26) Sklenak, S.; Dedecek, J.; Li, C.; Wichterlova, B.; Gabova, V.; Sierka, M.; Sauer, J. Phys. Chem. Chem. Phys. 2009, 11, 1237-1247.
(27) Dedecek, J.; Lucero, M. J.; Li, C. B.; Gao, F.; Klein, P.; Urbanova, M.; Tvaruzkova, Z.; Sazama, P.; Sklenak, S. J. Phys. Chem. C 2011, 115, 11056-11064.
(28) Sklenak, S.; Dedecek, J.; Li, C. B.; Gao, F.; Jansang, B.; Boekfa, B.; Wichterlova, B.; Sauer, J. Collect. Czech. Chem. Commun. 2008, 73, 909-920.
(29) Dedecek, J.; Sklenak, S.; Li, C.; Wichterlova, B.; Gabova, V.; Brus, J.; Sierka, M.; Sauer, J. J. Phys. Chem. C 2009, 113, 1447-1458.
(30) Dedecek, J.; Sklenak, S.; Li, C. B.; Gao, F.; Brus, J.; Zhu, Q. J.; Tatsumi, T. J. Phys. Chem. C 2009, 113, 14454-14466.
(31) Vjunov, A.; Fulton, J. L.; Huthwelker, T.; Pin, S.; Mei, D.; Schenter, G. K.; Govind, N.; Camaioni, D. M.; Hu, J. Z.; Lercher, J. A. J. Am. Chem. Soc. 2014, 136, 8296-8306.
(32) Holzinger, J.; Nielsen, M.; Beato, P.; Brogaard, R. Y.; Buono, C.; Dyballa, M.; Falsig, H.; Skibsted, J.; Svelle, S. J. Phys. Chem. C 2019, 123, 7831-7844.
(33) Kiricsi, I.; Flego, C.; Pazzuconi, G.; Parker, W. O.; Millini, R.; Perego, C.; Bellussi, G. J. Phys. Chem. 1994, 98, 4627-4634.
(34) Corma, A.; Garcia, H. Chem. Rev. 2002, 102, 3837-3892.
(35) Cejka, J.; Wichterlova, B. Catal. Rev. Sci. Eng. 2002, 44, 375-421.
(36) Corma, A. Chem. Rev. 1995, 95, 559-614.
(37) van Bokhoven, J. A.; van der Eerden, A. M. J.; Koningsberger, D. C. J. Am. Chem. Soc. 2003, 125, 7435-7442.
(38) Brus, J.; Kobera, L.; Schoefberger, W.; Urbanova, M.; Klein, P.; Sazama, P.; Tabor, E.; Sklenak, S.; Fishchuk, A. V.; Dedecek, J. Angew. Chem., Int. Ed. 2015, 54, 541-545.
(39) Kobera, L.; Dedecek, J.; Klein, P.; Tabor, E.; Brus, J.; Fishchuk, A. V.; Sklenak, S. J. Chem. Phys. 2022, 156, 104702.
(40) Sklenak, S.; Andrikopoulos, P. C.; Boekfa, B.; Jansang, B.; Novakova, J.; Benco, L.; Bucko, T.; Hafner, J.; Dedecek, J.; Sobalik, Z. J. Catal. 2010, 272, 262-274.
(41) Tabor, E.; Lemishka, M.; Sobalik, Z.; Mlekodaj, K.; Andrikopoulos, P. C.; Dedecek, J.; Sklenak, S. Commun. Chem. 2019, 2, 71.
(42) Sazama, P.; Moravkova, J.; Sklenak, S.; Vondrova, A.; Tabor, E.; Sadovska, G.; Pilar, R. ACS Catal. 2020, 10, 3984-4002.
(43) Tabor, E.; Dedecek, J.; Mlekodaj, K.; Sobalik, Z.; Andrikopoulos, P. C.; Sklenak, S. Sci. Adv. 2020, 6, eaaz9776.
(44) Dedecek, J.; Tabor, E.; Andrikopoulos, P. C.; Sklenak, S. Int. J. Quantum Chem. 2021, 121, e26611.
(45) Tabor, E.; Lemishka, M.; Olszowka, J. E.; Mlekodaj, K.; Dedecek, J.; Andrikopoulos, P. C.; Sklenak, S. ACS Catal. 2021, 11, 2340-2355.
(46) Jisa, K.; Novakova, J.; Schwarze, M.; Vondrova, A.; Sklenak, S.; Sobalik, Z. J. Catal. 2009, 262, 27-34.
(47) Panov, G. I.; Sobolev, V. I.; Kharitonov, A. S. J. Mol. Catal. 1990, 61, 85-97.
(48) Panov, G. I.; Kharitonov, A. S.; Sobolev, V. I. Appl. Catal., A 1993, 98, 1-20.
(49) Sobolev, V. I.; Dubkov, K. A.; Panna, O. V.; Panov, G. I. Catal. Today 1995, 24, 251-252.
(50) Dubkov, K. A.; Sobolev, V. I.; Talsi, E. P.; Rodkin, M. A.; Watkins, N. H.; Shteinman, A. A.; Panov, G. I. J. Mol. Catal., A 1997, 123, 155-161.
(51) Dubkov, K. A.; Ovanesyan, N. S.; Shteinman, A. A.; Starokon, E. V.; Panov, G. I. J. Catal. 2002, 207, 341-352.
(52) Yuan, S.; Li, Y. D.; Peng, J. Y.; Questell-Santiago, Y. M.; Akkiraju, K.; Giordano, L.; Zheng, D. J.; Bagi, S.; Roman-Leshkov, Y.; Shao-Horn, Y. Adv. Energy Mater. 2020, 10, 2002154.
(53) Mlekodaj, K.; Lemishka, M.; Sklenak, S.; Dedecek, J.; Tabor, E. Chem. Commun. 2021, 57, 3472-3475.
(54) Sklenak, S.; Groizard, T.; Jirglova, H.; Sazama, P.; Dedecek, J. J. Phys. Chem. C 2022, 126, 4854-4861.
(55) Primo, A.; Garcia, H. Chem. Soc. Rev. 2014, 43, 7548-7561.
(56) Vogt, E. T. C.; Weckhuysen, B. M. Chem. Soc. Rev. 2015, 44, 7342-7370.
(57) Roy, S.; Hegde, M. S.; Madras, G. Appl. Energy 2009, 86, 2283-2297.
(58) Shan, W. P.; Song, H. Catal. Sci. Technol. 2015, 5, 4280-4288.
(59) Beale, A. M.; Gao, F.; Lezcano-Gonzalez, I.; Peden, C. H. F.; Szanyi, J. Chem. Soc. Rev. 2015, 44, 7371-7405.
(60) Schwartz, T. J.; O'Neill, B. J.; Shanks, B. H.; Dumesic, J. A. ACS Catal. 2014, 4, 2060-2069.
(61) Ennaert, T.; Van Aelst, J.; Dijkmans, J.; De Clercq, R.; Schutyser, W.; Dusselier, M.; Verboekend, D.; Sels, B. F. Chem. Soc. Rev. 2016, 45, 584-611.
(62) Bhanja, P.; Bhaumik, A. Fuel 2016, 185, 432-441.
(63) Zakaria, Z.; Kamarudin, S. K. Renew. Sustain. Energy Rev. 2016, 65, 250-261.
(64) Narsimhan, K.; Iyoki, K.; Dinh, K.; Roman-Leshkov, Y. ACS Cent. Sci. 2016, 2, 424-429.
(65) Ravi, M.; Ranocchiari, M.; van Bokhoven, J. A. Angew. Chem., Int. Ed. 2017, 56, 16464-16483.
(66) Bian, Z. F.; Das, S.; Wai, M. H.; Hongmanorom, P.; Kawi, S. ChemPhysChem 2017, 18, 3117-3134.
(67) Catizzone, E.; Bonura, G.; Migliori, M.; Frusteri, F.; Giordano, G. Molecules 2018, 23, 31.
(68) Snyder, B. E. R.; Bols, M. L.; Schoonheydt, R. A.; Sels, B. F.; Solomon, E. I. Chem. Rev. 2018, 118, 2718-2768.
(69) Derouane, E. G.; Vedrine, J. C.; Pinto, R. R.; Borges, P. M.; Costa, L.; Lemos, M.; Lemos, F.; Ribeiro, F. R. Catal. Rev. Sci. Eng. 2013, 55, 454-515.
(70) Serrano, D. P.; Melero, J. A.; Morales, G.; Iglesias, J.; Pizarro, P. Catal. Rev. Sci. Eng. 2018, 60, 1-70.
(71) Dedecek, J.; Capek, L.; Kaucky, D.; Sobalik, Z.; Wichterlova, B. J. Catal. 2002, 211, 198-207.
(72) Benco, L.; Bucko, T.; Grybos, R.; Hafner, J.; Sobalik, Z.; Dedecek, J.; Hrusak, J. J. Phys. Chem. C 2007, 111, 586-595.
(73) Mortier, W. J. J. Phys. Chem. 1977, 81, 1334-1338.
(74) Schlenker, J. L.; Pluth, J. J.; Smith, J. V. Mater. Res. Bull. 1978, 13, 169-174.
(75) Dalconi, M. C.; Cruciani, G.; Alberti, A.; Ciambelli, P.; Rapacciuolo, M. T. Microporous Mesoporous Mater. 2000, 39, 423-430.
(76) Dalconi, M. C.; Alberti, A.; Cruciani, G. J. Phys. Chem. B 2003, 107, 12973-12980.
(77) Dalconi, M. C.; Alberti, A.; Cruciani, G.; Ciambelli, P.; Fonda, E. Microporous Mesoporous Mater. 2003, 62, 191-200.
(78) Van der Borght, K.; Batchu, R.; Galvita, V. V.; Alexopoulos, K.; Reyniers, M. F.; Thybaut, J. W.; Marin, G. B. Angew. Chem., Int. Ed. 2016, 55, 12817-12821.
(79) Shi, J.; Wang, Y.; Yang, W.; Tang, Y.; Xie, Z. Chem. Soc. Rev. 2015, 44, 8877-8903.
(80) Kubanek, P.; Wichterlova, B.; Sobalik, Z. J. Catal. 2002, 211, 109-118.
(81) Yahiro, H.; Iwamoto, M. Appl. Catal., A 2001, 222, 163-181.
(82) Brandenberger, S.; Krocher, O.; Tissler, A.; Althoff, R. Appl. Catal., B 2010, 95, 348-357.
(83) Gramm, F.; Baerlocher, C.; McCusker, L. B.; Warrender, S. J.; Wright, P. A.; Han, B.; Hong, S. B.; Liu, Z.; Ohsuna, T.; Terasaki, O. Nature 2006, 444, 79-81.
(84) Hong, S. B.; Min, H. K.; Shin, C. H.; Cox, P. A.; Warrender, S. J.; Wright, P. A. J. Am. Chem. Soc. 2007, 129, 10870-10885.
(85) Danilina, N.; Payrer, E. L.; van Bokhoven, J. A. Chem. Commun. 2010, 46, 1509-1510.
(86) Portilla, M. T.; Llopis, F. J.; Martinez, C.; Valencia, S.; Corma, A. Appl. Catal., A 2011, 393, 257-268.
(87) Min, H. K.; Hong, S. B. J. Phys. Chem. C 2011, 115, 16124-16133.
(88) Min, H. K.; Cha, S. H.; Hong, S. B. ACS Catal. 2012, 2, 971-981.
(89) Xu, W.; Miller, S. J.; Agrawal, P. K.; Jones, C. W. Appl. Catal., A 2013, 459, 114-120.
(90) Cha, S. H.; Lee, K.; Byun, Y.; Hong, S. B. J. Phys. Chem. C 2016, 120, 11552-11560.
(91) Byun, Y.; Cha, S. H.; Jeon, H. J.; Hong, S. B. J. Phys. Chem. C 2016, 120, 6125-6135.
(92) Bleken, F.; Skistad, W.; Barbera, K.; Kustova, M.; Bordiga, S.; Beato, P.; Lillerud, K. P.; Svelle, S.; Olsbye, U. Phys. Chem. Chem. Phys. 2011, 13, 2539-2549.
(93) Franch-Marti, C.; Alonso-Escobar, C.; Jorda, J. L.; Peral, I.; Hernandez-Fenollosa, J.; Corma, A.; Palomares, A. E.; Rey, F.; Guilera, G. J. Catal. 2012, 295, 22-30.
(94) Moreno-Gonzalez, M.; Blasco, T.; Gora-Marek, K.; Palomares, A. E.; Corma, A. Catal. Today 2014, 227, 123-129.
(95) Wang, Y. A.; Gao, Y.; Chu, W. F.; Zhao, D. P.; Chen, F. C.; Zhu, X. X.; Li, X. J.; Liu, S. L.; Xie, S. J.; Xu, L. Y. J. Mater. Chem. A 2019, 7, 7573-7580.
(96) Xu, H.; Zhu, J.; Zhu, L. F.; Zhou, E. N.; Shen, C. Molecules 2020, 25, 3722.
(97) Liu, X. H.; Liao, Z. F.; Wang, H. L.; Fu, J. L.; Zheng, J. B.; Zhang, N. W.; Chen, B. H. Appl. Catal., A 2022, 642, 118676.
(98) Park, S. J.; Jang, H. G.; Lee, K. Y.; Cho, S. J. Microporous Mesoporous Mater. 2018, 256, 155-164.
(99) Ham, H.; Xuan, N. T.; Jung, H. S.; Kim, J.; Roh, H. S.; Bae, J. W. Catal. Today 2021, 369, 112-122.
(100) Swies, A.; Kowalczyk, A.; Gil, B.; Chmielarz, L. RSC Adv. 2022, 12, 9395-9403.
(101) Jung, H. S.; Xuan, N. T.; Bae, J. W. Microporous Mesoporous Mater. 2021, 310, 110669.
(102) Chen, Z. Q.; Liu, S. Y.; Zhang, H. K.; He, P.; Ren, J.; Wen, X. D.; Li, Y. W. Catal. Sci. Technol. 2021, 11, 2094-2102.
(103) Nie, Q.; Si, J. Q.; Meng, C.; Lan, T.; Sun, W. D.; Zhao, G. F.; Liu, Y.; Lu, Y. ACS Sustain. Chem. Eng. 2022, 10, 9282-9294.
(104) Dapsens, P. Y.; Mondelli, C.; Perez-Ramirez, J. Chem. Soc. Rev. 2015, 44, 7025-7043.
(105) Song, S.; Yuen, V. F. K.; Di, L.; Sun, Q. M.; Zhou, K.; Yan, N. Angew. Chem., Int. Ed. 2020, 59, 19846-19850.
(106) Capek, L.; Dedecek, J.; Sazama, P.; Wichterlova, B. J. Catal. 2010, 272, 44-54.
(107) Narayanan, S.; Tamizhdurai, P.; Mangesh, V. L.; Ragupathi, C.; Krishnan, P. S.; Ramesh, A. RSC Adv. 2021, 11, 250-267.
(108) US 8426633, 2013.
(109) Boronat, M.; Martinez-Sanchez, C.; Law, D.; Corma, A. J. Am. Chem. Soc. 2008, 130, 16316-16323.
(110) Boronat, M.; Martinez, C.; Corma, A. Phys. Chem. Chem. Phys. 2011, 13, 2603-2612.
(111) Alayon, E. M.; Nachtegaal, M.; Ranocchiari, M.; van Bokhoven, J. A. Chem. Commun. 2012, 48, 404-406.
(112) Alayon, E. M. C.; Nachtegaal, M.; Kleymenov, E.; van Bokhoven, J. A. Microporous Mesoporous Mater. 2013, 166, 131-136.
(113) Alayon, E. M. C.; Nachtegaal, M.; Bodi, A.; van Bokhoven, J. A. ACS Catal. 2014, 4, 16-22.
(114) Deka, U.; Juhin, A.; Eilertsen, E. A.; Emerich, H.; Green, M. A.; Korhonen, S. T.; Weckhuysen, B. M.; Beale, A. M. J. Phys. Chem. C 2012, 116, 4809-4818.
(115) Deka, U.; Lezcano-Gonzalez, I.; Weckhuysen, B. M.; Beale, A. M. ACS Catal. 2013, 3, 413-427.
(116) Lomachenko, K. A.; Borfecchia, E.; Negri, C.; Berlier, G.; Lamberti, C.; Beato, P.; Falsig, H.; Bordiga, S. J. Am. Chem. Soc. 2016, 138, 12025-12028.
(117) Heo, I.; Sung, S.; Park, M. B.; Chang, T. S.; Kim, Y. J.; Cho, B. K.; Hong, S. B.; Choung, J. W.; Nam, I.-S. ACS Catal. 2019, 9, 9800-9812.
(118) Zhu, Q.; Kondo, J. N.; Tatsumi, T.; Inagaki, S.; Ohnuma, R.; Kubota, Y.; Shimodaira, Y.; Kobayashi, H.; Domen, K. J. Phys. Chem. C 2007, 111, 5409-5415.
(119) Gallego, E. M.; Li, C. G.; Paris, C.; Martin, N.; Martinez-Triguero, J.; Boronat, M.; Moliner, M.; Corma, A. Chem. Eur. J. 2018, 24, 14631-14635.
(120) Ferri, P.; Li, C. G.; Millan, R.; Martinez-Triguero, J.; Moliner, M.; Boronat, M.; Corma, A. Angew. Chem., Int. Ed. 2020, 59, 19708-19715.
(121) Adiga, S.; Aebi, D.; Bryce, D. L. Can. J. Chem. 2007, 85, 496-505.
(122) Bucko, T.; Hafner, J.; Benco, L. J. Chem. Phys. 2004, 120, 10263-10277.
(123) Benco, L.; Bucko, T.; Hafner, J.; Toulhoat, H. J. Phys. Chem. B 2005, 109, 22491-22501.
(124) Hafner, J.; Benco, L.; Bucko, T. Top. Catal. 2006, 37, 41-54.
(125) Grybos, R.; Hafner, J.; Benco, L.; Toulhoat, H. J. Phys. Chem. C 2007, 111, 6454-6464.
(126) Grybos, R.; Hafner, J.; Benco, L.; Raybaud, P. J. Phys. Chem. C 2008, 112, 12349-12362.
(127) Benco, L.; Bucko, T.; Grybos, R.; Hafner, J.; Sobalik, Z.; Dedecek, J.; Sklenak, S.; Hrusak, J. J. Phys. Chem. C 2007, 111, 9393-9402.
(128) Andrikopoulos, P. C.; Sobalik, Z.; Novakova, J.; Sazama, P.; Sklenak, S. ChemPhysChem 2013, 14, 520-531.
(129) Kresse, G.; Hafner, J. Phys. Rev. B 1993, 48, 13115-13118.
(130) Kresse, G.; Hafner, J. Phys. Rev. B 1994, 49, 14251-14269.
(131) Kresse, G.; Furthmuller, J. Phys. Rev. B 1996, 54, 11169-11186.
(132) Kresse, G.; Furthmuller, J. Comput. Mater. Sci. 1996, 6, 15-50.
(133) Kresse, G.; Joubert, D. Phys. Rev. B 1999, 59, 1758-1775.
(134) Balabanov, N. B.; Peterson, K. A. J. Chem. Phys. 2006, 125, 074110.
(135) Kachurovskaya, N. A. J. Phys. Chem. B 2004, 108, 5944-5950.
(136) Li, G. N.; Pidko, E. A.; van Santen, R. A.; Feng, Z. C.; Li, C.; Hensen, E. J. M. J. Catal. 2011, 284, 194-206.
(137) Goltl, F.; Hafner, J. J. Chem. Phys. 2012, 136, 064501.
(138) Goltl, F.; Hafner, J. J. Chem. Phys. 2012, 136, 064502.
(139) Goltl, F.; Hafner, J. J. Chem. Phys. 2012, 136, 064503.
(140) Li, G. N.; Pidko, E. A.; van Santen, R. A.; Li, C.; Hensen, E. J. M. J. Phys. Chem. C 2013, 117, 413-426.
(141) VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Comput. Phys. Commun. 2005, 167, 103-128.
(142) Chung, L. W.; Sameera, W. M. C.; Ramozzi, R.; Page, A. J.; Hatanaka, M.; Petrova, G. P.; Harris, T. V.; Li, X.; Ke, Z. F.; Liu, F. Y.; Li, H. B.; Ding, L. N.; Morokuma, K. Chem. Rev. 2015, 115, 5678-5796.
(143) Eichler, U.; Kolmel, C. M.; Sauer, J. J. Comput. Chem. 1997, 18, 463-477.
(144) Sierka, M.; Sauer, J. J. Chem. Phys. 2000, 112, 6983-6996.
(145) Sierka, M.; Sauer, J. Faraday Discuss. 1997, 106, 41-62.
(146) Frisch, M. J. T., G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A., et al. Gaussian 09, Gaussian, Inc.: Wallingford, CT, 2011.
(147) Wolinski, K.; Hinton, J. F.; Pulay, P. J. Am. Chem. Soc. 1990, 112, 8251-8260.
(148) Haser, M.; Ahlrichs, R. J. Comput. Chem. 1989, 10, 104-111.
(149) Treutler, O.; Ahlrichs, R. J. Chem. Phys. 1995, 102, 346-354.
(150) Eichkorn, K.; Treutler, O.; Ohm, H.; Haser, M.; Ahlrichs, R. Chem. Phys. Lett. 1995, 240, 283-289.
(151) Eichkorn, K.; Treutler, O.; Ohm, H.; Haser, M.; Ahlrichs, R. Chem. Phys. Lett. 1995, 242, 652-660.
(152) Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Theor. Chem. Acc. 1997, 97, 119-124.
(153) Gale, J. D. J. Chem. Soc., Faraday Trans. 1997, 93, 629-637.
(154) Gale, J. D.; Rohl, A. L. Mol. Simul. 2003, 29, 291-341.
(155) Whittleton, S. R.; Vicente, A.; Fernandez, C.; Rastegar, S. F.; Fishchuk, A. V.; Sklenak, S. Microporous Mesoporous Mater. 2018, 267, 124-133.
(156) Schafer, A.; Huber, C.; Ahlrichs, R. J. Chem. Phys. 1994, 100, 5829-5835.
(157) Jensen, F. J. Chem. Theory Comput. 2008, 4, 719-727.
(158) Steinmann, S. N.; Corminboeuf, C. J. Chem. Theory Comput. 2011, 7, 3567-3577.
(159) Steinmann, S. N.; Corminboeuf, C. J. Chem. Phys. 2011, 134, 044117.
(160) Grimme, S. J. Comput. Chem. 2004, 25, 1463-1473.
(161) Mortier, W. J. Zeolites 1982, 38, 1-67.
(162) Yakimov, A. V.; Ravi, M.; Verel, R.; Sushkevich, V. L.; van Bokhoven, J. A.; Coperet, C. J. Am. Chem. Soc. 2022, 144, 10377-10385.
(163) Attfield, M. P.; Weigel, S. J.; Cheetham, A. K. J. Catal. 1997, 172, 274-280.
(164) Dalconi, M. C.; Cruciani, G.; Alberti, A.; Ciambelli, P. Catal. Today 2005, 110, 345-350.
(165) Pickering, I. J.; Maddox, P. J.; Thomas, J. M.; Cheetham, A. K. J. Catal. 1989, 119, 261-265.
(166) Snyder, B. E. R.; Vanelderen, P.; Bols, M. L.; Hallaert, S. D.; Bottger, L. H.; Ungur, L.; Pierloot, K.; Schoonheydt, R. A.; Sels, B. F.; Solomon, E. I. Nature 2016, 536, 317-321.
(167) Kitajima, N.; Fujisawa, K.; Fujimoto, C.; Morooka, Y.; Hashimoto, S.; Kitagawa, T.; Toriumi, K.; Tatsumi, K.; Nakamura, A. J. Am. Chem. Soc. 1992, 114, 1277-1291.
(168) Sharma, V. K. Coord. Chem. Rev. 2013, 257, 495-510.